PRM Conductor Quick Reference
PRM Conductor is an application that aids in the creation of targeted MS methods. This program filters "bad" transitions, and selects "good" peptides via a set of criteria, and helps the user to create one or more instrument methods that will be acquired with at least a required number of points per peak.
Launching PRM Conductor from the Thermo tools menu will open the application. If there was data in the Skyline document, a report will be exported from Skyline and imported to PRM Conductor. The view will look similar to below.
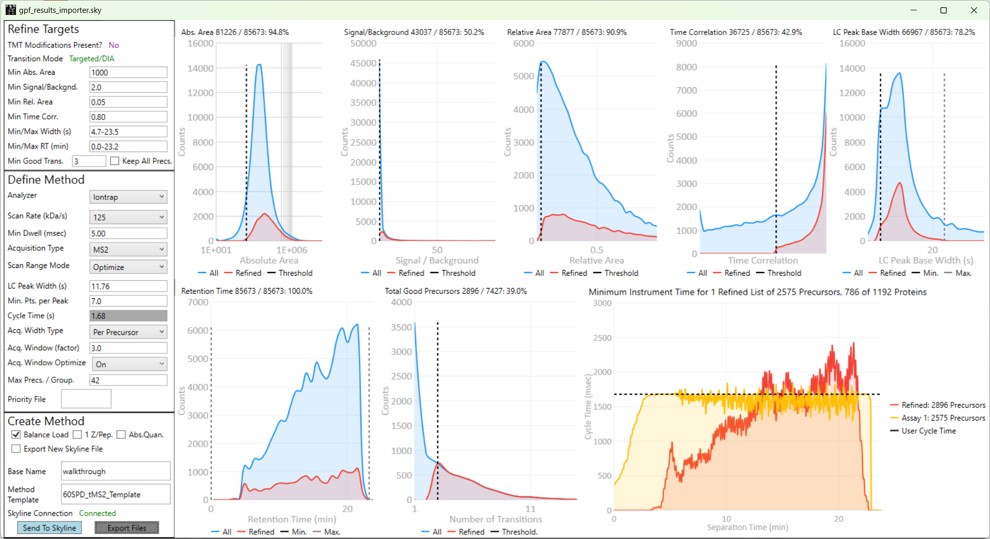
The user parameters are split into 3 sections; Refine Targets, Define Method, and Create Method
Refine Targets
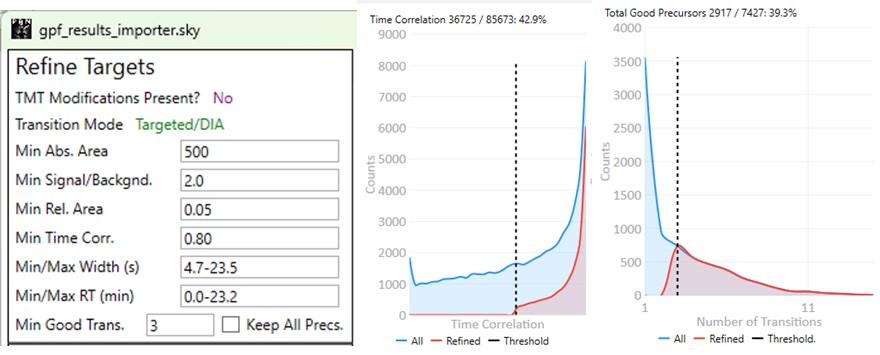
These parameters configure a series of simple filters for the transitions, and allows to filter precursors by the number of good transitions that they have.
- TMT Modifications Present?
- This value is No if there are no TMT modifications. If TMT modifications are present, then the exported methods can account for this, for example by choosing MS3 precursors that have TMT tags.
- Transition Mode
- This value is Precursor/DDA if ALL the analytes have precursor transitions. The 1.0 PRM Conductor version would set this mode if ANY precursor transition was present, which was confusing. This DDA mode means that MS2 data may only be acquired once per LC peak, and so some metrics like Time Correlation are not valid. This mode is common for Small Molecule analysis when data are imported from Compound Discoverer. PRM Conductor is still useful in this case, to create instrument methods, but it may be advisable to select Keep All Precs. option, and apply filtering on a later targeted data set.
- This value is usally Targeted/DIA, enabling the full filtering functionality of the Time Correlation filter.
- Min. Abs. Area
- Filters transitions by requiring that they have a minimum absolute area value. This value may have to change for data acquired on different instruments. For example, the intensity scaling on Orbitrap instruments is arbitrarily scaled to a value that is ~10x the value for Ion trap and Astral data.
- Min. Signal / Backgnd.
- Filters transitions by requiring that they have a minimum signal to background ratio. Signal is the Skyline peak area, and Background is the Skyline background value. This value should be used sparingly, because the current background metric that we use is not as good as it could be.
- Min. Rel. Area
- Filters transitions by requiring that they have a minimum relative area to the largest transition for its precursor.
- Min. Time Corr.
- Filters transitions by requiring that they have a minimum correlation to the median transition. This value is called Shape Correlation by Skyline,and was defined by Searle et. al., and later in some detail by Heil et. al.
- Min./Max. Width (s)
- Filters transitions by requiring that their base LC width be within the given bounds. The bounds is computed from the Skyline FWHM and converted to a base peak width by assuming a Gaussian shape and multiplying by 2.54. See the supplement of Remes et. al. for a derivation.
- Min./Max. RT (min)
- Filters precursors by requiring that their retention time be within the given bounds.
- Min. Good Transitions
- Filters precursors by requiring at least this many transitions that satisfy the above filters.
- Keep All Precs.
- This option ensures that no precursors are filtered, and the top Min. Good Transitions are kept. If there are not enough good transitions, "bad" transitions are selected according to Area x Time Corr. This option is useful when the user wants to keep all the precursors and just clean up the transitions, or is using DDA mode.
Define Method
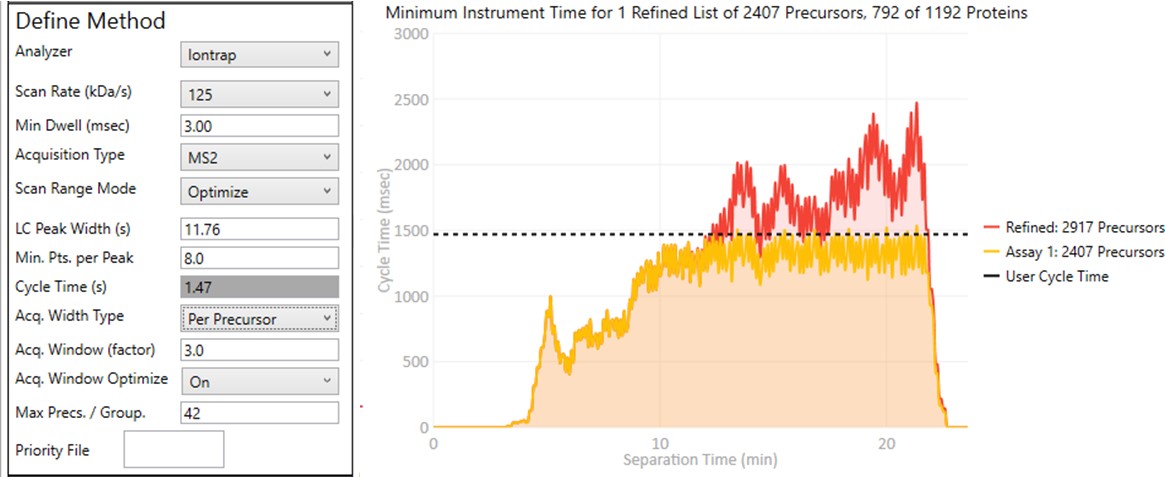
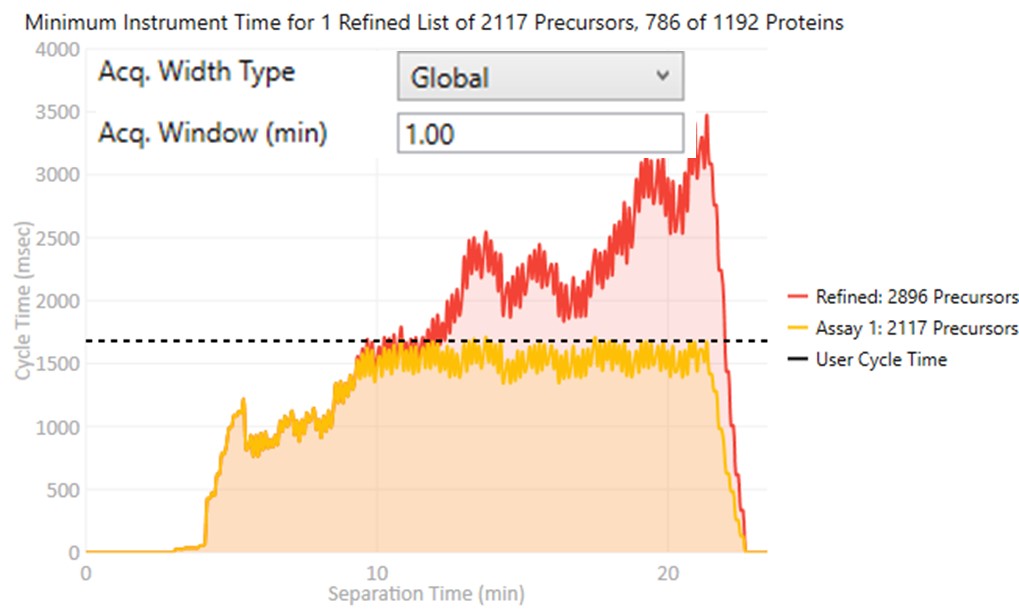
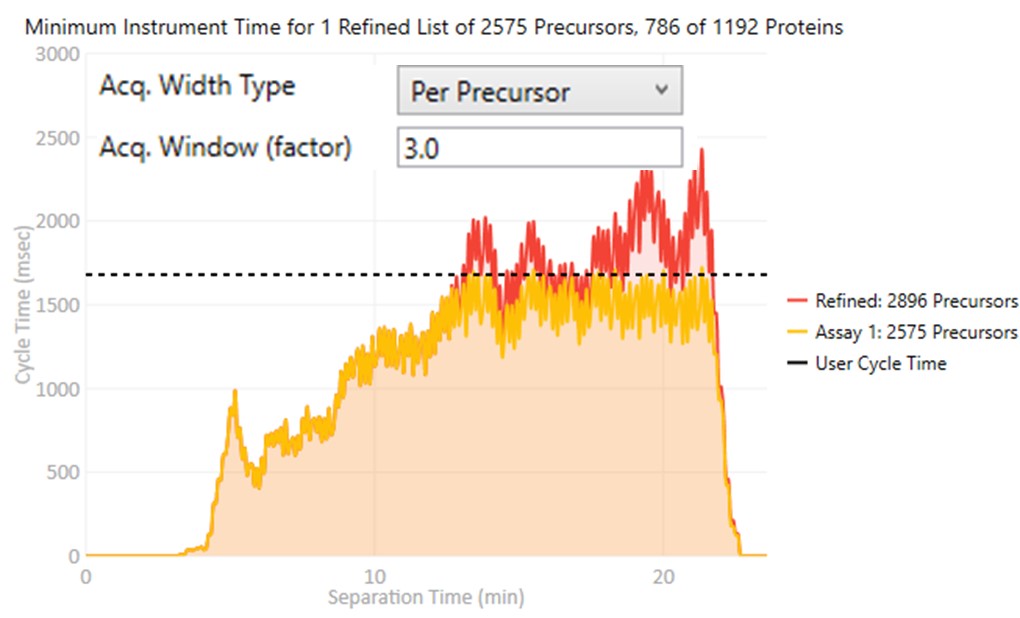
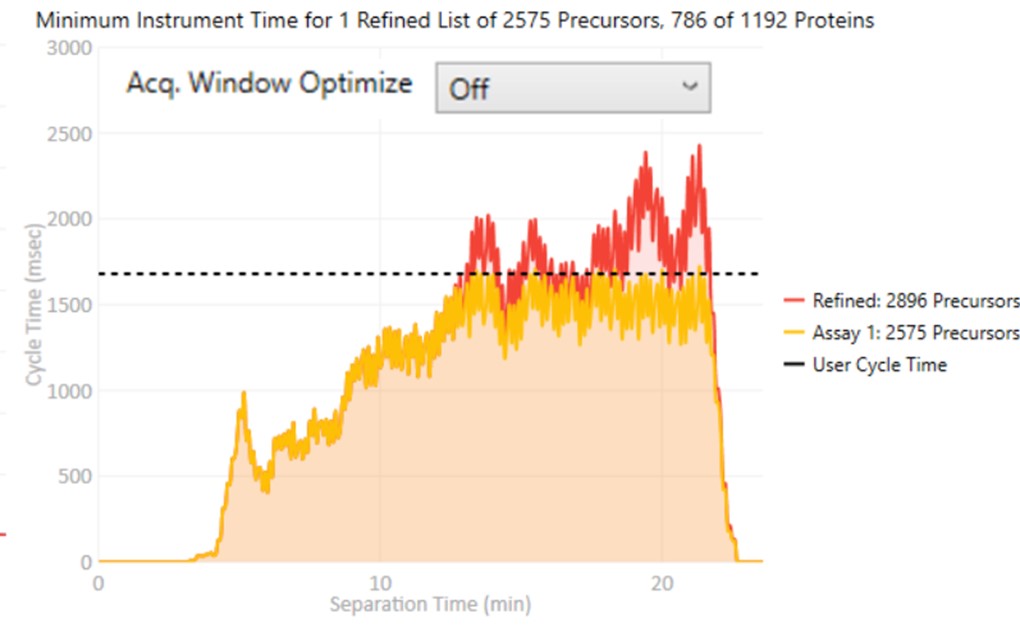
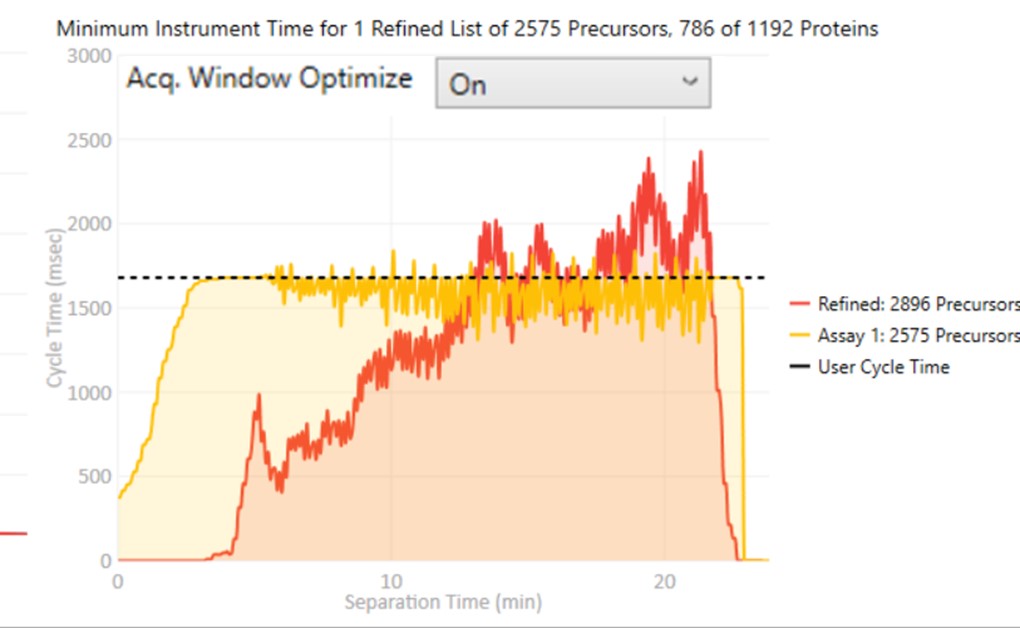
These parameters control how many targets can be included in the assay. When parameters in this section are changed, and the 'enter' key is pressed, the graph on the right will update, showing how many precursors can be included in the assay, while respecting Cycle Time defined by the LC Peak Width and the Min. Pts. per Peak.
Analyzer
- Ion Trap
- Scan Rate (kDa/s)
- Sets the analysis scan rate. This value affects the acquisition rate, and therefore the assay target capacity.
-
Astral
- Currently the Astral has no analyzer-specific parameters
-
Orbitrap
- Resolution (k)
- Sets the analysis resolution. This value affects the acquisition rate, and therefore the assay target capacity.
-
Triple
- Max #Transitions
- Only up to this number of transitions will be selected, since this value linearly decreases the assay target capacity.

-
Min Dwell (msec)
- Also called injection time on many instruments, this value sets the smallest amount of precursor accumulation time per target. Typically we set this value small enough so that it does not affect the assay capacity, and rely on the dynamic AGC algorithm to boost the injection time according to the available time in the cycle. See Remes et. al. for a description of the dynamic AGC process.
- In some cases, the experimenter may know that to achieve high quality data, a minimum dwell time is needed, and will adjust this value accordingly.
Acquisition Type
- MS2
- The standard analysis type, where each acquisition contains data for a single precursor that is mass selected and fragmented.
- MS3
- Currently only available when Ion Trap analyzer is selected, in this mode, each acquisition contains data for a single precursor that is mass selected, fragmented, and one or more fragments are further mass selected and fragmented.
- The Max # Transitions parameter becomes available in this mode, limiting the number of MS3 precursors that will be simultaneous mass analyzed in the second MS stage. For purposes of the acquisition rate estimations, this mode currently assumes that resonance CID with 4 msec activation time is used for the first activation, and HCD is used for the second activation. In reality if the user selects a method template the uses resonance CID for the second activation, then the activation for each MS3 precursor is performed serially, taking at least #transitions x activation_time to be performed.
-
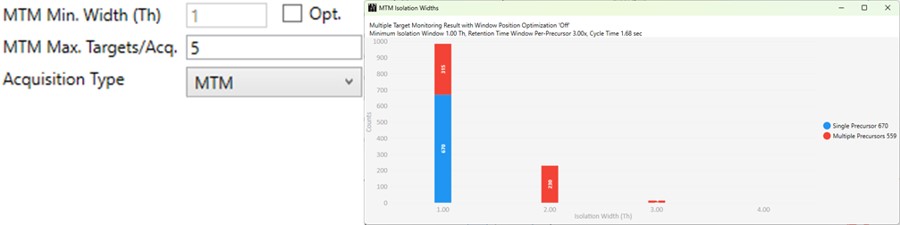
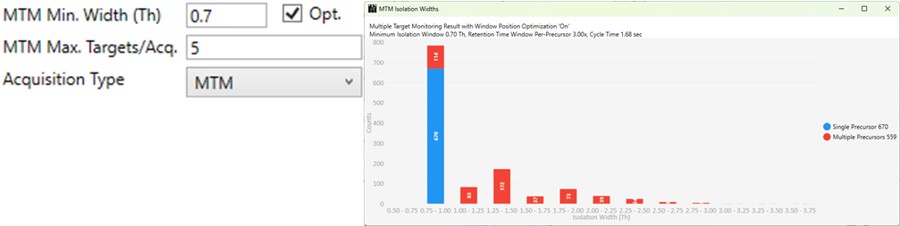
MTM
- Multiple target monitoring is a type of method that can be generated from 1 Th isolation window GPF DIA data. It analyzes the GPF data to determine when it is safe to expand the number of targets, or the isolation width, to encompass multiple targets. MTM can be used to expand the number of targets in an assay compared to normal PRM, or alternatively for the same number of targets, MTM can enable the use faster gradients and higher throughput, or higher injection times and better sensitivity.
- When the Opt. check box is unchecked, then the isolation widths are multiples of the GPF isolation width, which is shown in a disabled text box (1 here). A pop-up window shows the distribution of isolation widths, and how many of them are for single or multiple targets.
- When the Opt. check box is checked, then the isolation widths are customized for each acquisition, such that the window size is (largest_mz - smallest_mz) + MTM Min. Width (Th).
-
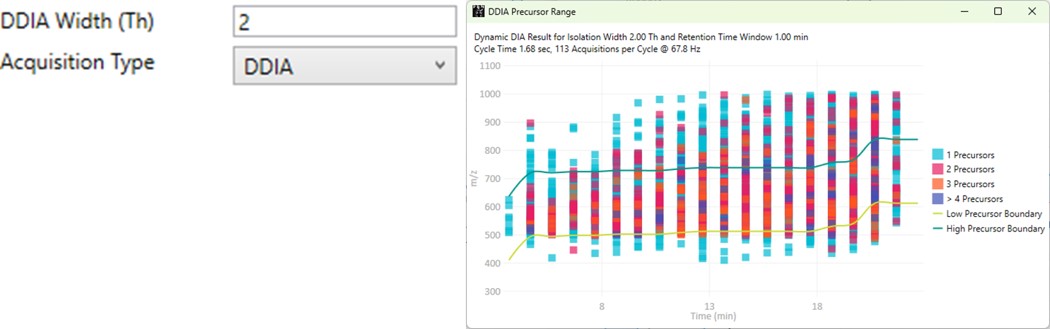
DDIA
- Dynamic DIA is a type of DIA method in which the precursor range shifts as a function of the experiment time. This method can be used to acquire data with a narrower isolation window than would be possible if data for the entire precursor range had to acquired on each cycle.
- The DDIA Width(Th) sets the isolation width that will be used. Narrower widths will acquire higher quality data, for fewer precursors.
- A pop-up plot appears that shows the density of precursors in the Skyline document, along with lines that trace the proposed precursor range as a function of experiment time.
Scan Range Mode
-
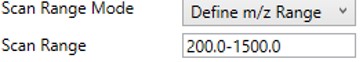
Define m/z Range
- Define an explicit first and last mass for the acquired spectra. The acquisition rate is inversely proportional to the size of the m/z range.
-
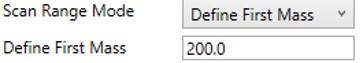
Define First Mass
- Define a constant first mass for all acquisitions. The last mass is determined based on the charge state, such that last_mass = precursor_mz * charge + 10
-
Auto
- The first and last mass are determined automatically. The last mass is determined as in the Define First Mass mode. The first mass is determined as the lowest mass that still retains most of the ions at the last mass, based on experimental evaluations.
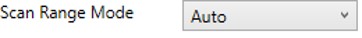
-
Optimize
- The first and last mass are set based on the set of "good" transitions for a precursor plus a small buffer. This mode enables the fastest acquisition rates possible for the Ion trap analysis, where the mass analysis time is directly proportional to the scan range.
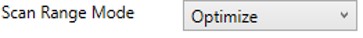
Other Define Method Parameters
Create Method
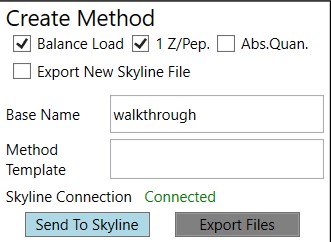
These parameters control aspects of how the method will be created.

 prm_conductor_quick_ref4.gif
prm_conductor_quick_ref4.gif prm_conductor_quick_ref5.gif
prm_conductor_quick_ref5.gif