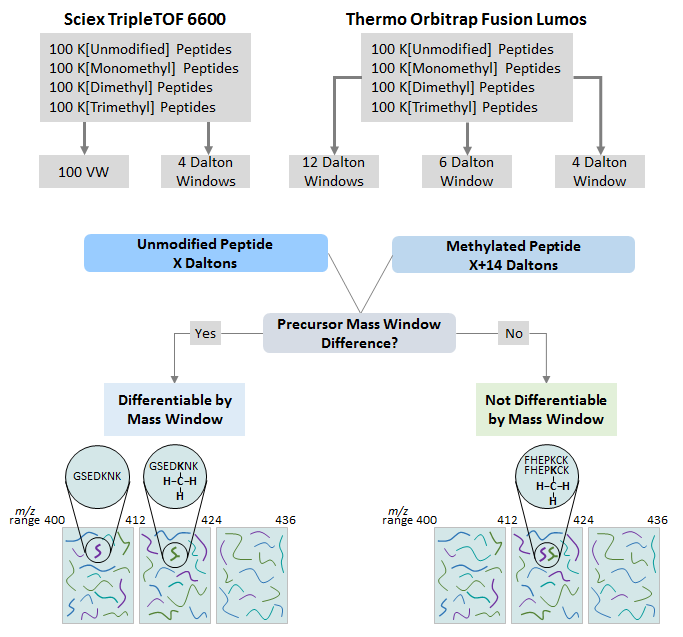
To establish DIA-MS parameters to allow for differentiating a methylated peptide from an unmodified peptide, first a DDA-MS library was built on a Sciex 6600 Triple TOF based on the DDA acquisition of a pool of 400 synthesized peptides (400 femtomoles/peptide) containing either K[Unmodified], K[Monomethyl], K[Dimethyl], or K[Trimethyl]. DIA acquisitions of 400 femtomoles/peptide of the methyl peptide pool were then acquired using a 4 Dalton precursor mass windows and 100 variable mass windows. A DDA library was also created from the methyl peptide pool acquired on the Orbitrap Fusion Lumos (200 femtomoles/peptide). DIA acquisitions of 200 femtomoles/peptide were then acquired with 4, 6, and 12 Dalton fixed precursor mass windows respectively. Each acquisition contains a pool consisting of 100 synthesized unmodified and mono-, di- and tri-methylated peptides. Peptides were resuspended in 0.1% Formic Acid and pooled at 1:1 ratio.