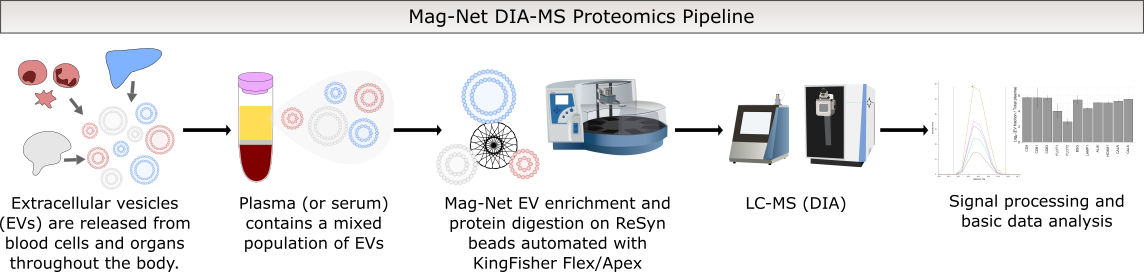
The MacCoss laboratory is currently offering a number of quantitative proteomics assays as a service. Included in these assays is the recently developed Mag-Net plasma assay using LC-MS. This can be done using small quantities of plasma or serum across multiple species. Click here to contact us and get the process started!
Our group has extensive experience applying quantitative proteomics to the analysis of biofluids. Additionally, we have been leaders in the development and application of data independent acquisition-mass spectrometry and targeted proteomics methods.
We offer the expertise to develop and validate customized multiplex targeted protein assays. We have developed a workflow based on gas phase fractionation-data independent acquisition to expedite the selection of target peptides that provide robust quantitative measurements directly within the sample matrix. We can go from candidate list to scheduled peptide assay in a short time pending the peptides are above the limit of detection. All assays are developed using:
If you have a question that isn't addressed in the above sections, check out our FAQs. For additional questions, please feel free to email us at services@maccosslab.org.
|
Showing: all announcements
|
||
| Use of a hybrid quadrupole mass filter - radial ejection linear ion trap and intelligent data acquisition for multiplex targeted proteomics | ||
| 2024-06-10 12:46:25 | ||
 Stellar MS 2D Diagram.png Stellar MS 2D Diagram.png | ||
| view announcement | ||
| Methodology describing the use of data independent acquisition to develop targeted methods | ||
| 2024-06-01 22:56:03 | ||
| view announcement | ||
| 2024 ASMS | ||
| 2024-06-01 19:36:02 | ||
 2024-ASMS MacCoss Lab.jpeg 2024-ASMS MacCoss Lab.jpeg | ||
| view announcement | ||
| A framework for quality control in quantitative proteomics | ||
| 2024-06-01 19:33:03 | ||
| view announcement | ||
| Preprint describing the Mag-Net plasma enrichment strategy up on Biorxiv | ||
| 2024-06-01 19:32:00 | ||
| view announcement | ||