A Peptide-Centric Quantitative Proteomics Dataset for the Phenotypic Assessment of Alzheimer’s Disease
Merrihew GE, Park J, Plubell D, Searle BC, Keene CD, Larson EB, Bateman R, Perrin RJ, Chhatwal JP, Farlow MR, McLean CA, Ghetti B, Newell KL, Frosch MP, Montine TJ, MacCoss MJ. A peptide-centric quantitative proteomics dataset for the phenotypic assessment of Alzheimer's disease. Sci Data. 2023 Apr 14;10(1):206. doi: 10.1038/s41597-023-02057-7. PMID: 37059743; PMCID: PMC10104800.
- Organism: Homo sapiens
- Instrument: Orbitrap Fusion Lumos
- SpikeIn:
No
- Keywords:
Quantitative Proteomics, DIA Mass Spectrometry, Alzheimer’s disease, peptide-centric, human brain tissue
-
Lab head: Michael MacCoss
Submitter: Gennifer Merrihew
Alzheimer’s disease (AD) is a looming public health disaster with limited interventions. Desperately needed progress in therapeutic development rests on more detailed understanding of molecular pathogenesis. The vast majority of existing data for AD pathogenesis in humans is from standard neuropathologic assessments, e.g., neuritic plaques and neurofibrillary tangles or biochemical measurement of a limited number of analytes directly related to neuropathologic features, e.g., amyloid-β peptides and hyperphosphorylated tau. Standard neuropathologic evaluation is highly valuable as the only tool that provides comprehensive assessment of diseases that afflict an individual brain, but it also is severely limited in its ability to investigate molecular pathogenesis. Advances in mass spectrometry-based proteomics techniques provide robust quantitative measures, allowing for insight into the protein phenotype in brains with AD neuropathology. Careful selection of individuals in multiple research cohorts enabled us, for the first time, to investigate the full spectrum of molecular signatures specific to autosomal dominant AD dementia (ADD), sporadic ADD with minimal comorbidities, individuals without dementia who had high histopathologic burden of AD, and cognitively normal individuals with no or minimal AD histopathologic burden.
Two 25 μm frozen sections of brain tissue were resuspended in 120 μl of lysis buffer of 5% SDS, 50mM triethylammonium bicarbonate (TEAB), 2mM MgCl2, 1X HALT phosphatase and protease inhibitors, vortexed and and briefly sonicated at setting 3 for 10 s with a Fisher sonic dismembrator model 100. A microtube was loaded with 30 μl of lysate and capped with a micropestle for homogenization with a Barocycler 2320EXT (Pressure Biosciences Inc.) for a total of 20 minutes at 35°C with 30 cycles of 20 seconds at 45,000 psi followed by 10 seconds at atmospheric pressure. Protein concentration was measured with a BCA assay. Homogenate of 50 μg was added to a process control of 800 ng of yeast enolase protein (Sigma) which was then reduced with 20 mM DTT and alkylated with 40 mM IAA. Lysates were then prepared for S-trap column (Protifi) binding by the addition of 1.2% phosphoric acid and 350 μl of binding buffer (90% Methanol, 100 mM TEAB). The acidified lysate was bound to column incrementally, followed by 3 wash steps with binding buffer to remove SDS and 3 wash steps with 50:50 methanol:chloroform to remove lipids and a final wash step with binding buffer. Trypsin (1:10) in 50mM TEAB was then added to the S-trap column for digestion at 47°C for one hour. Hydrophilic peptides were then eluted with 50 mM TEAB and hydrophobic peptides were eluted with a solution of 50% acetonitrile in 0.2% formic acid. Elutions were pooled, speed vacuumed and resuspended in 0.1% formic acid.
Injection of samples are one ug of total protein (16 ng of enolase process control) and 150 fmol of a heavy labeled Peptide Retention Time Calibrant (PRTC) mixture (Pierce). The PRTC is used as a peptide process control. Library pools are an equivalent amount of every sample (including references) in the batch. For example, a batch library pool consists of the 14 samples from the batch and two references. System suitability (QC) injections are 150 fmol of PRTC and BSA.
One µg of each sample with 150 femtomole of PRTC was loaded onto a 30 cm fused silica picofrit (New Objective) 75 µm column and 3.5 cm 150 µm fused silica Kasil1 (PQ Corporation) frit trap loaded with 3 µm Reprosil-Pur C18 (Dr. Maisch) reverse-phase resin analyzed with a Thermo Easy-nLC 1200. The PRTC mixture is used to assess system suitability before and during analysis. Four of these system suitability runs are analyzed prior to any sample analysis and then after every six sample runs another system suitability run is analyzed. Buffer A was 0.1% formic acid in water and buffer B was 0.1% formic acid in 80% acetonitrile. The 40-minute system suitability gradient consists of a 0 to 16% B in 5 minutes, 16 to 35% B in 20 minutes, 35 to 75% B in 1 minute, 75 to 100% B in 5 minutes, followed by a wash of 9 minutes and a 30 minute column equilibration. The 110-minute sample LC gradient consists of a 2 to 7% for 1 minutes, 7 to 14% B in 35 minutes, 14 to 40% B in 55 minutes, 40 to 60% B in 5 minutes, 60 to 98% B in 5 minutes, followed by a 9 minute wash and a 30 minute column equilibration. Peptides were eluted from the column with a 50°C heated source (CorSolutions) and electrosprayed into a Thermo Orbitrap Fusion Lumos Mass Spectrometer with the application of a distal 3 kV spray voltage. For the system suitability analysis, a cycle of one 120,000 resolution full-scan mass spectrum (350-2000 m/z) followed by a data-independent MS/MS spectra on the loop count of 76 data-independent MS/MS spectra using an inclusion list at 15,000 resolution, AGC target of 4e5, 20 millisecond (ms) maximum injection time, 33% normalized collision energy with a 8 m/z isolation window. For the sample digest, first a chromatogram library of 6 independent injections is analyzed from a pool of all samples within a batch. For each injection a cycle of one 120,000 resolution full-scan mass spectrum with a mass range of 100 m/z (400-500 m/z, 500-600 m/z, 600-700 m/z, 700-800 m/z, 800-900 m/z, 900-1000 m/z) followed by a data-independent MS/MS spectra on the loop count of 26 at 30,000 resolution, AGC target of 4e5, 60 ms maximum injection time, 33% normalized collision energy with a 4 m/z overlapping isolation window. The chromatogram library data is used to quantify proteins from individual sample runs. These individual runs consist of a cycle of one 120,000 resolution full-scan mass spectrum with a mass range of 350-2000 m/z, AGC target of 4e5, 100 ms maximum injection time followed by a data-independent MS/MS spectra on the loop count of 76 at 15,000 resolution, AGC target of 4e5, 20 ms maximum injection time, 33% normalized collision energy with an overlapping 8 m/z isolation window. Application of the mass spectrometer and LC solvent gradients are controlled by the ThermoFisher Xcalibur (version 3.1.2412.24) data system.
Brain tissue samples were stratified into 4 groups based on clinical, pathological and genetic data and four brain regions (superior and middle temporal gyri or SMTG, Hippocampus at the level of the lateral geniculate nucleus, inferior parietal lobule or IPL and Caudate nucleus at the level of the anterior commissure). Cognitive status was determined as dementia or not dementia by DSM-IVR criteria. Individuals diagnosed as not dementia were from the Adult Changes in Thought (ACT) study and were included only if the last research evaluation was within 2 years of death and the last cognitive screening (CASI) score was in the upper quartile for the ACT cohort (>90); our definition of HCF. Brains from individuals with HCF who had no or low ADNC were designated “HCF/low ADNC” and those with intermediate or high ADNC were designated “HCF/high ADNC''. All individuals diagnosed with ADD had intermediate or high level ADNC and were further subclassified as sporadic (“Sporadic ADD”) or ADD caused by a mutation in PSEN1, PSEN2, or APP (“Autosomal Dominant ADD”). Sporadic AD cases were from the ACT study and the University of Washington (UW) AD Research Center (ADRC), and Autosomal Dominant ADD cases were from the UW ADRC and the Dominantly Inherited Alzheimer Network (DIAN). Excluded was any case with LBD or LATE-NC other than involving amygdala, territorial infarcts, more than 2 cerebral microinfarcts, or hippocampal sclerosis. Time from death to cryopreservation of tissue, postmortem interval (PMI), was <8 hr in all cases except for those in the Autosomal Dominant ADD group. Details of sample stratification for the 4 brain regions (SMTG, Hippocampus, IPL and Caudate) are provided.
Each human brain region was divided into batches of 14 individual samples and 2 pooled references for a total of 16. The first batch of each region was also used to create a region-specific reference pool to be used as a “common reference” and/or single point calibrant, which was homogenized, aliquoted, frozen, and used to compare between batches within a brain region. Human cerebellum and occipital lobe tissue was homogenized, pooled, aliquoted and frozen to be used as a “batch reference” for comparison between batches and other brain regions. Batch design was randomly balanced based on group ratios. For example, batches from the SMTG brain region contained 5 “Sporadic ADD”, 4 “Autosomal Dominant ADD”, 2 “HCF/low ADNC”, and 3 “HCF/high ADNC” samples. Metadata for the samples from the Hippocampus, IPL and Caudate brain regions is provided.
Created on 6/15/22, 1:17 AM

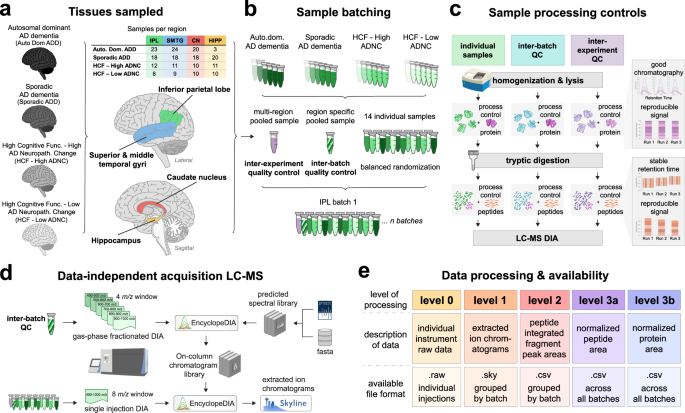 Figure1.png
Figure1.png