Table of Contents |
guest 2024-11-02 |
Validation with Subset Replicates
Analysis of Final Assay Replicates
Label Free - E. coli
E. coli Assay Development Overview
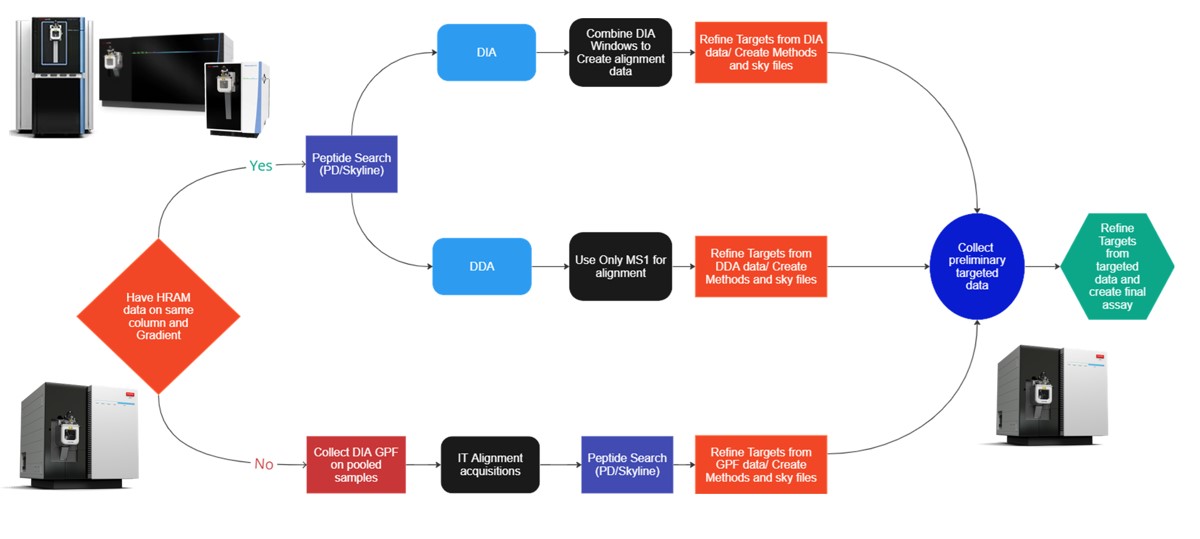
This tutorial will show you how to create a label-free targeted assay from discovery results. As shown in the figure below, there are two main routes to creating this kind of assay, to main difference being whether the discovery data comes from a high resolution accurate mass instrument like Astral, Exploris, or a Tribrid, or whether the discovery data comes from a Stellar. Once we have the discovery results in Skyline, the steps are largely the same. In this tutorial we'll look at an assay created from Stellar MS gas phase fractionation results.
Collect Gas Phase Fractionation Data on Pooled Samples
Overview of the Library Methods
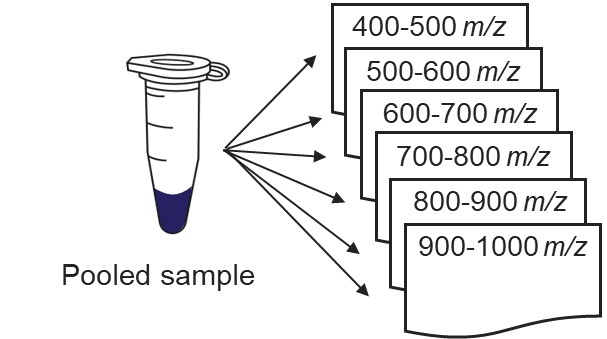
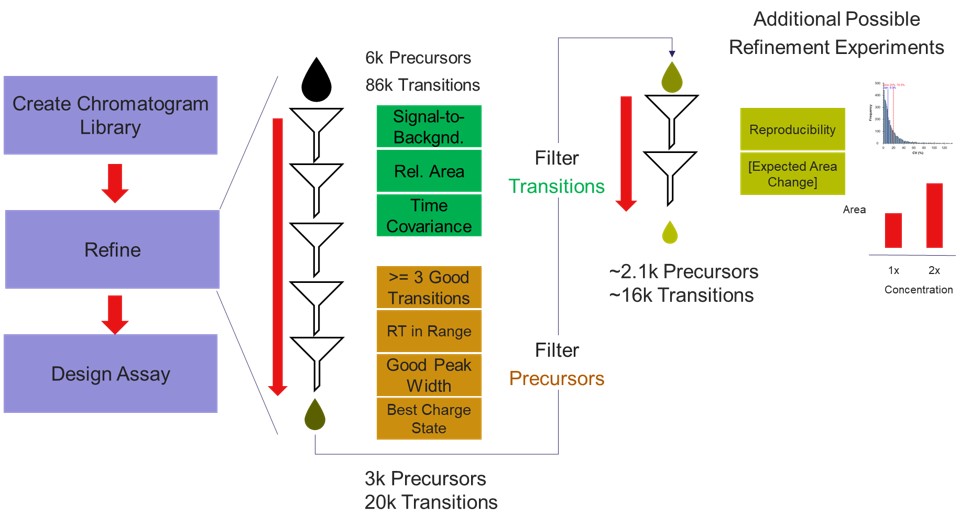
Our recommended technique for creating targeted assays starts with acquiring a set of narrow isolation window DIA data on a pooled or characteristic sample, creating what the MacCoss lab has termed a “chromatogram library”. This technique allows for a deep characterization of the detectable peptides in the sample, and the resulting data can be used to create a Skyline transition list of high quality targets. As outlined in in the figure below, generation of the library typically involves several (~6) LC injections, where each injection is focused on the analysis of a small region of precursor mass-to-charge. This technique has also been called “gas-phase fractionation” (GPF) to contrast with the traditional technique of off-line fraction collection and analysis. The GPF experiment should be performed with the same LC gradient as the final targeted assay, which allows us to use the library data for real-time retention time alignment during later tMS2 experiments.
Assuming that a pooled sample is available for analysis, the first step is to create a template instrument method file (.meth). Open the Instrument Setup program from Xcalibur, or the Standalone Method Editor (no LC drivers) if using a Workstation installation.
C:\Program Files\Thermo Scientific\Instruments\TNG\Thorium\1.0\System\Programs\TNGMethodEditor.exe
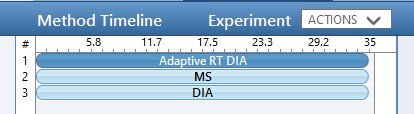
Use the method editor to open the file at Step 1. DIA GPF/60SPD_DIA_1Th_GPF.meth. This method includes 3 experiments:
- An Adaptive DIA experiment for retention time alignment that has a wide isolation window and ultrafast scan rate. We use Window Optimization Off so that later these windows will not be imported into Skyline, which can be accomplished because both the analytical DIA experiment (experiment #3) and later targeted methods will not use integer precursor m/z values, and Skyline will use a 0.0001 Th tolerance.
- An MS experiment, used for diagnostic purposes. Some DIA processing algorithms may also use the MS data for peptide searching, but currently nominal mass MS data don’t appear to be useful for DIA. Note that MS data can be used for data normalization in Skyline, even when MS data are not being imported.
- An analytical DIA experiment for peptide searching that use a narrow (~1 Th) isolation window. Window Optimization is set to “On” so that isolation window edges are in “forbidden” m/z zones and we don’t have to use any window overlap.
Assuming that the user has configured their LC driver with the Instrument Configuration tool, the LC driver options would appear as a tab in the pane on the left-hand side of the method, where the user has to design the length and type of gradient program to be used. Of course, the choice of gradient length is of great importance as it determines the number of compounds that can be analyzed and the experimental throughput. A general rule of thumb is that Stellar can analyze on the order of 5000 peptides in an hour gradient, where this number depends on how the peptide retention times are distributed and other factors and settings that will be explored later in the tutorial. The user will need to make sure that the MS part of the experiment has the same Method and Experiment Durations as the LC gradient.
While we are making methods, we should create a tMS2 method template with the same LC parameters as the library method file. One can start with Step 1. DIA GPF/60SPD_tMS2_Template.meth. This method has the same first two experiments as the GPF template, and the 3rd experiment been replaced with a tMSn experiment. Our software tool will later fill in the targeted table information in a new version of this file, as well as update the LC peak width and cycle time parameters and embed the reference data for the Adaptive RT.
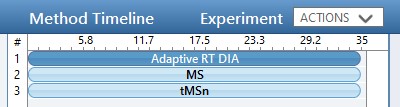
One thing to note about the targeted template is that the Dynamic Time Scheduling is set to Adaptive RT. When we save the method, a message tells us that the file is invalid because no reference file has been specified. This is okay, because PRM Conductor will fill in this information for us later.

Creating the GPF Methods
Once the GPF method template file is created and saved, the user should select that file with the GPF Creator tool. The tool will create a set of new methods based on this template, one for each precursor mass-to-charge region. The number of method files to be created depends on the settings in the user interface. The default precursor range for the tool is 400 to 1000 Th, as most tryptic peptides have a mass-to-charge ratio in this range. We recommend, for Stellar, to use an isolation width of 1 Th, which allows the DIA data to be searched even with traditional peptide search tools like SEQUEST and allows for a good determination of which transitions will be interference free in the tMS2 experiment. However some users have used 2 Th windows, in order to user slower scan rates and/or more injection time per scan without having to acquire 2x as many GP fractions. If the experiment Maximum Injection Time Mode is set to Auto, then the maximum injection time is the largest value that does not slow down acquisition, which depends on the Scan Rate. For this simple tool, we assume a scan range of 200-1500 Th. The largest amount of injection time and the slowest acquisition rate is afforded by the 33 kDa/s scan rate, and the smallest amount of injection time and the fastest scan rate is afforded by the 200 kDa/s scan rate.
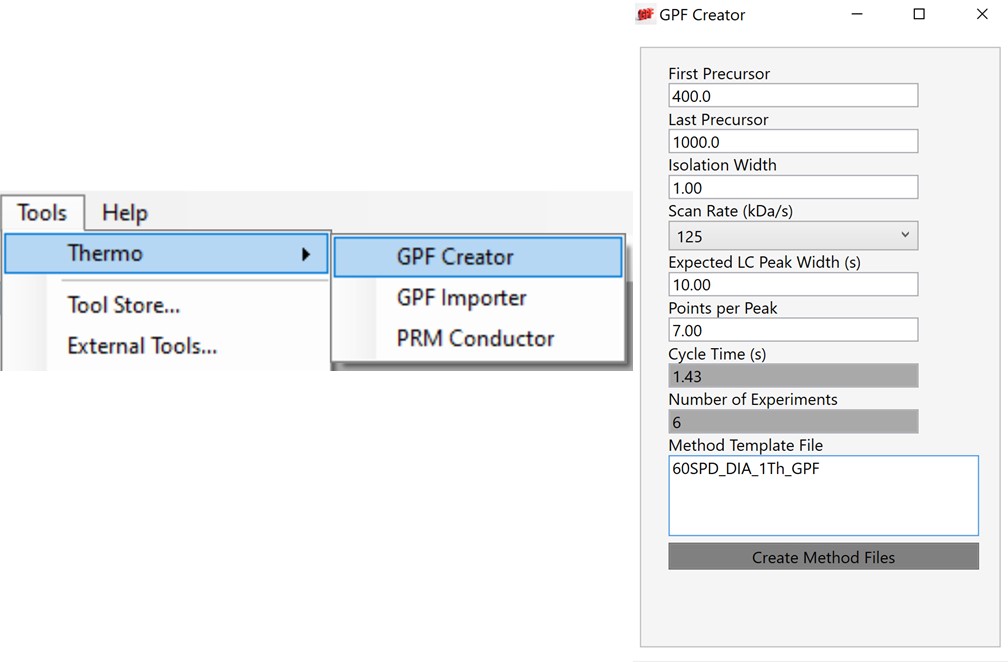
For most applications, we prefer to use 67 or 125 kDa/s and set the Maximum Injection Time Mode to Dynamic as in the figure below, which lets the maximum injection time scale with the time available in the cycle.
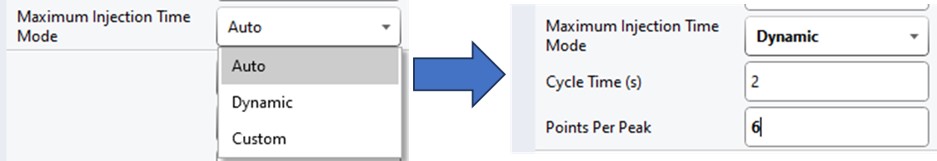
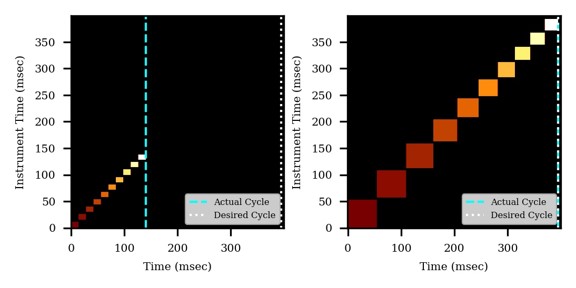
If the Cycle Time is set to 2 seconds and the cycles take less than 2 seconds, then the remaining time in the cycle is distributed to the acquisitions inversely proportionally to the intensity of the precursors, ex. less intense precursors get more injection time. In this figure, the left panel shows a hypothetical situation at the start of a cycle. We calculate the minimum amount of time that each of the acquisitions needs, and sum this to get the blue, vertical, dashed line. The remaining time in the cycle is the white, vertical, dotted line. Each acquisition is colored, with the most intense precursors colored white, based on previous data. In the right panel, the amount of time the acquisitions actually take is displayed, where the less intense precursors were given more injection time.
The expected peak width in GPF Creator is the LC peak width at the base in seconds. This value can be determined by checking a few peaks in a quality control (QC) experiment, for example an injection of the Pierce Retention Time Calibration standard. The LC peak width combined with the desired number of sampling points across the peak width, the isolation width, and the Scan Rate determines the number of LC injections needed to create the library.
Once the parameters are set, the user selects the Create Method Files button, which will create the instrument method files needed for the library. The user can create a sequence in Xcalibur and run these methods along with any blanks and QC’s that they want. We have collected data for a mix of 200 ng/ul E. coli with 200 ng/ul HeLa, and placed the resulting raw files in Step 1. DIA GPF\Raw.
Search Library against Organism Database
The next step is to search the data to find candidate peptide targets. Ion trap DIA data with 1 Th isolation windows can be searched for peptides using Proteome Discoverer and the SEQUEST or CHIMERYS search engines. We have included example templates for Proteome Discoverer with this tutorial. SEQUEST in Proteome Discoverer was built to handle data dependent acquisition experiments, and so a few parameters are set to unexpected values. The Precursor Mass Tolerance is set to 0.75 Da, which means that each spectrum is queried against the peptides in a window that is +/- 0.75 * charge_state. For the spectrum selector node, we set Unrecognized Charge Replacements to 2; 3, so that each spectrum is searched as both charge state 2 and 3, because Stellar DIA acquisitions are marked with Charge 0, which is Unrecognized for PD. This practice, plus the wide precursor mass tolerance, means that each spectrum in the chromatogram library takes a long time to search; on the order of an hour for a 30 minute gradient method using a standard instrument PC, ex 6 hours to search a GPF experiment. CHIMERYS searching is often much faster for this kind of experiment.
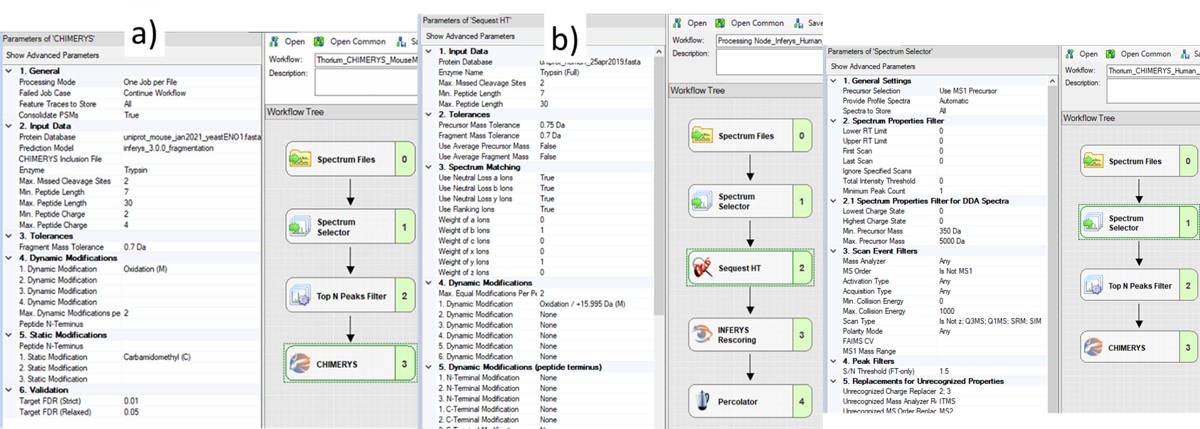
Also, in the spectrum selector node we set the Scan Type to “Is Not Z” (only Full), so that the large isolation width Adaptive RT acquisitions are filtered, which have a Z in the scan header.
A nice, new feature in the Proteome Discoverer 3.1 SP1 is the addition of the Consolidate PSMs option in the Chimerys 1. General section. Setting this parameter to True tells Chimerys to keep on the best PSM per peptide/charge precursor. This consolidation has the effect of speeding up the Consensus workflow significantly, as well as reducing output file size and the amount of time needed for Skyline to import the results.
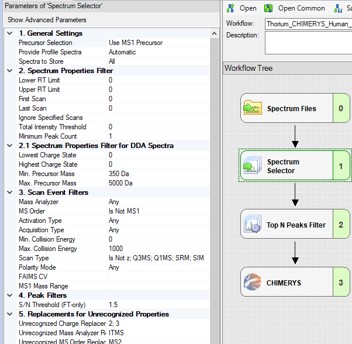
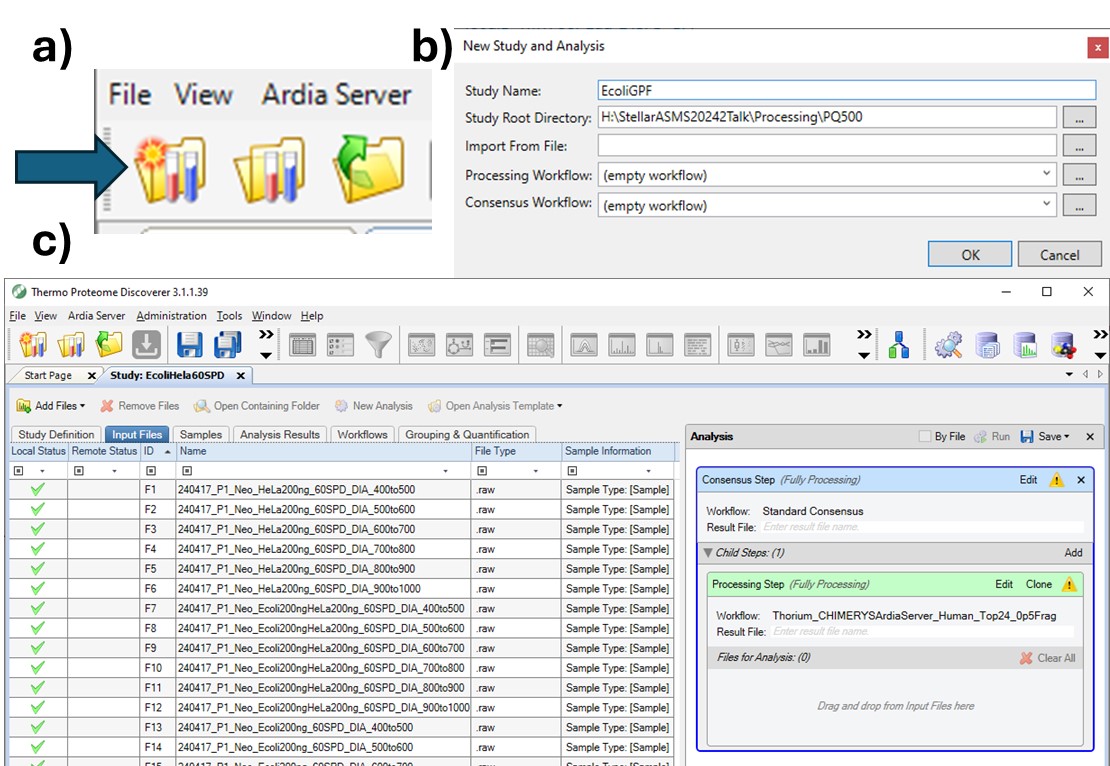
To process the files in Proteome Discoverer, one can first create a study (a), set the study name and directory, and the processing and consensus workflows (b). The workflows can also be set or modified once the study has been created. When the study has been created, one uses Add Files to choose the library raw files, and drags them to the Files for Analysis region, and then presses the Run button to start the analysis (c)
Manual Import Search Results into Skyline
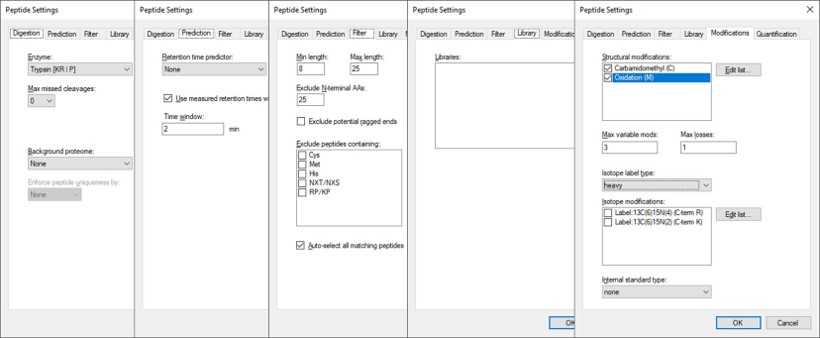
After Proteome Discoverer finishes processing the data, a file is produced with the .pdResult extension, and another one with the .pdResultDetails extension. Both files must be in the same folder for Skyline to import the search results and to start refining a First-Draft targeted method. We will first show you how to import the results manually, and then we will present a simple tool to perform the import with the settings that we recommend for ion trap analysis. To import the PD results manually, create a new Skyline document, and save it as ImportedGPFResults\gpf_results_manual.sky. Open Settings \ Peptide Settings, and set the parameters as in the figure below. The main points are to ensure that no Library is selected, and the Modifications do not specify any isotope modifications. Skyline will check for Structural modifications in the imported search results and ask you about any that are not already specified. Press Okay on the Peptide Settings dialog box.
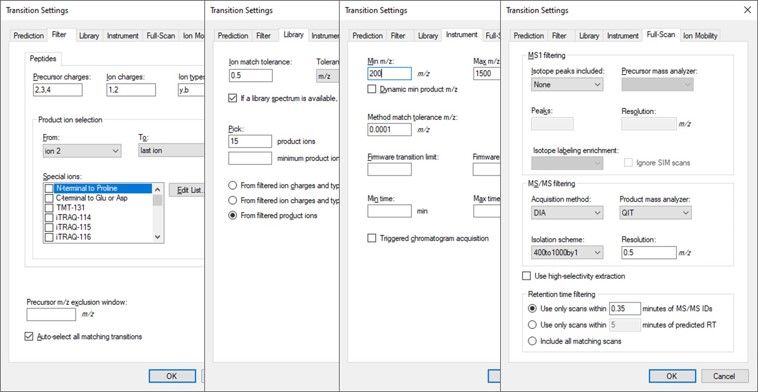
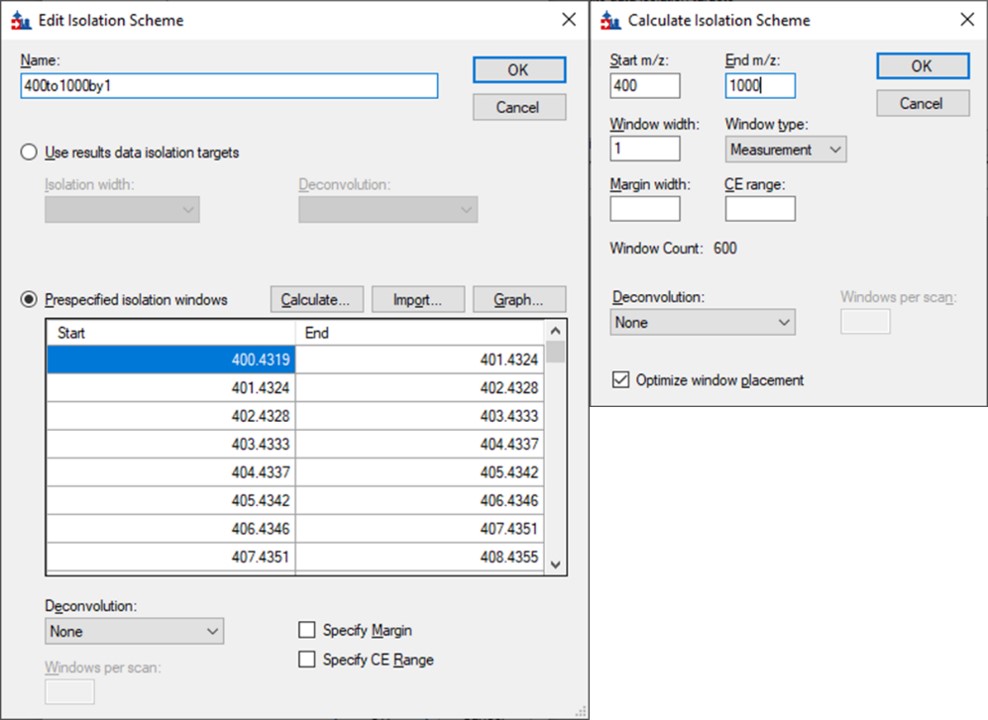
Open the Settings \ Transition Settings and use settings like below. Set Precursor charges to 2, 3, 4, Ion charges to 1,2, and Ion types to y,b. Don’t specify ‘p’ in Ion types, because PRM Conductor currently takes this as a sign that DDA is being used and loses some functionality. Set Product ion selection from ion 2 to last ion. For Ion match tolerance use 0.5 m/z, with a maximum of 15 product ions, filtered from product ions. Set the Min m/z to 200, and Max m/z to 1500 (whatever was used for acquiring the GPF data), with Method match tolerance of 0.0001. Set MS1 filtering to None, and MS/MS filtering to Acquisition method DIA, Product mass analyzer QIT with Resolution 0.5. For the Isolation scheme, if a suitable scheme is not yet created, select Add. Here we set 0.35 for the Retention time filtering, but a wider window may be more acceptable for longer gradients.
As below, once Add is selected for Isolation Scheme, give it a suitably descriptive name, then press Calculate. Set the Start and End m/z to the limits and isolation width used in the GPF experiment, and click the Optimize window placement button, assuming you used that option in the GPF experiment. Press Ok twice to close the Isolation Scheme Editor, and once more to close the Transition Settings.
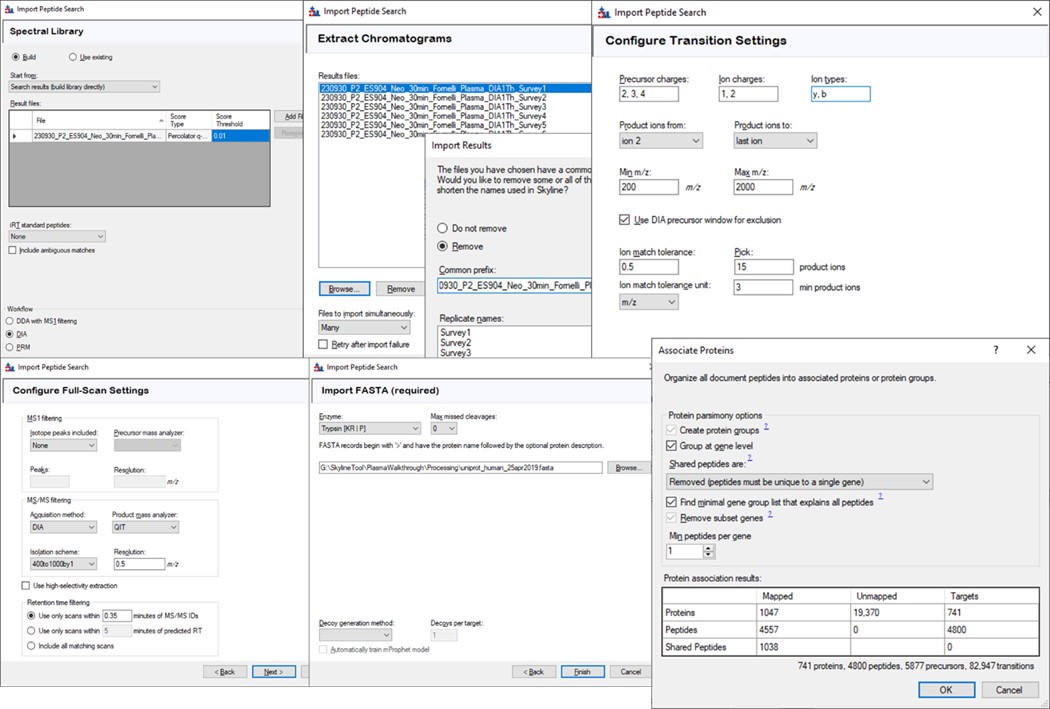
Save your Skyline document at this point, and select File / Import / Peptide Search. Use the Add Files button to navigate to and select the Step 1. DIA GPF/Processing/240417_P1_Neo_Ecoli200ngHeLa200ng_60SPD_DIA.pdResult file.
As in Figure below, select Score Threshold 0.01, Workflow DIA and press Next. Skyline will build a .blib file from the .pdResult, which will take several minutes. If you get an error during this step, ensure that the .pdResultDetails file is also present in the same folder as the .pdResult file, for PD >= 3.1, and ensure that you are using an updated version of Skyline (preferably the Daily version). When this step finishes, Skyline presents a .raw file chooser dialog, select Browse and select the files in Raw / GPF. Press Next, then you can remove or not remove portions of the file names to shorten them, and press Okay. The next Add Modifications tab should be empty if the Peptide Settings / Modifications were already specified, so press Next again. In the Configure Transition Settings, Skyline will have overwritten several of your Transition settings, so remove the ‘p’ from Ion types, and set Ion match tolerance to 0.5, and Max product ions 15, Min 3 and press Next. In the Configure Full Scan Settings, set the MS1 Isotope Peaks to None, and press Next. In the Import FASTA tool, Browse and select the .fasta file in the Processing folder. You can set the Decoy generation to None, especially if you used PSM Consolidation to reduce to a single PSM per peptide, then press Finish. Skyline will start associating peptides to proteins now, displaying a progress bar, and eventually shows an Associate Proteins tool. Here the most strict setting would be to create protein groups and remove any peptides that are not unique to a protein, but alternatively one might choose to Assign to the first gene, for example. Press Okay, and Skyline will import the results from the .raw file, which may take a few minutes. Note that one could have skipped the results importing and pressed cancel at any time after the library was created. The library would be available to browse with View / Spectral Libraries, and the peptides could be added to the document with Add All. Occasionally this process can be used if there were any errors or issues with modifications. Additionally one could use this strategy to load the .raw files as a single multi-injection replicate, the way that the GPF Importer will do, which makes viewing the results much more convenient as we’ll see below. When the results are finished loading, save the gpf_results_manual.sky file.
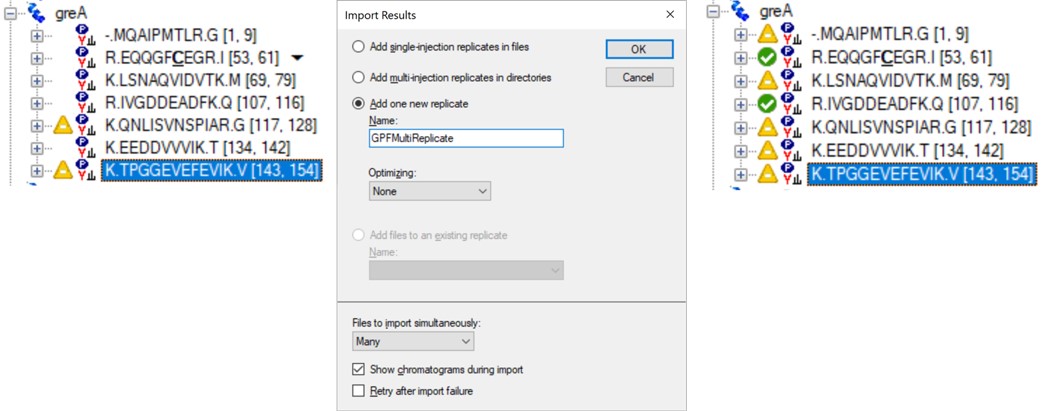
Click on the second gene labeled greA to expand it. Notice that with the 600to700 file selected, only two peptides are visible. With the scheme we have used, each .raw file was loaded on its own, instead of as part of a whole. To see the difference, use Edit / Manage Results (or Ctrl+R) to open the Manage Results tool, press Remove All and Okay. Now use File / Import / Results. When asked if you want to use decoys say No. In the Import Results tool, select Add one new replicate and use the name GPFMultiReplicate, and press Okay. Choose the six .raw files in the Raw / GPF folder again, and they will be immediately imported if you had finished the importing with the Wizard. The greA gene that is expanded now will have all its peptides completely filled in, like below. Save the gpf_results_manual.sky document.
GPF Import Search Results into Skyline
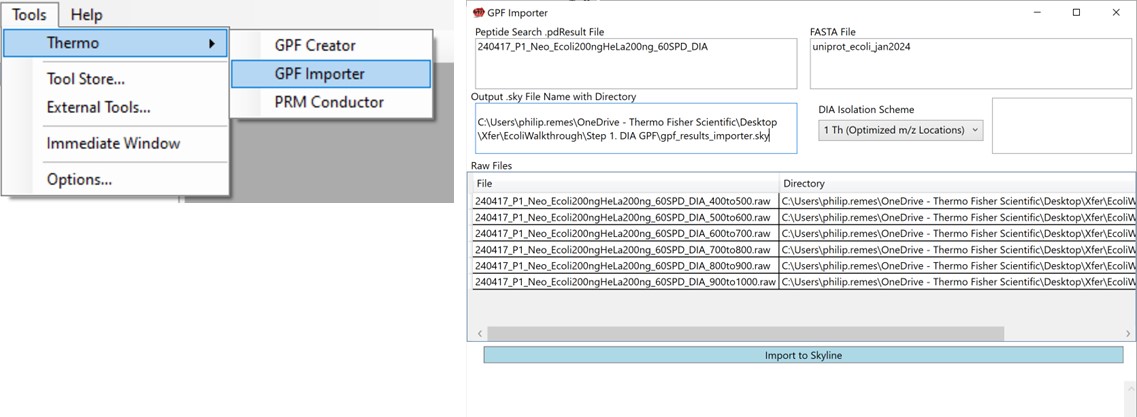
We’ll now look at how the GPF Importer does the same thing as above, but with fewer steps. To use the GPF Importer tool use Tools / Thermo / GPF Importer. Double click the Peptide Search and FASTA file boxes to select the .pdResult file and FASTA file respectively. In the Output file Path, Copy/Paste in the output directory you want, and type an appropriate name for the new Skyline file. Double-click the Raw File pane and select the associated GPF .raw files and press the Import to Skyline button. The log window shows the progress of the import, which takes several minutes. When it finishes, the result will be the same as in the figure below when the multi-injection replicate was imported.
This is the end of the first step. We have Skyline files with peptide search results in them. In the next steps we'll refine the results on our way to making a final assay.
Validation with Subset Replicates
- Refining the Search Results
Refining the Search Results
Use PRM Conductor Skyline plugin to refine the transitions and precursors
Launching PRM Conductor
Once the results are imported into Skyline, the next step is to create a First-Draft version of the targeted assay using the PRM Conductor Skyline plugin. If you followed the steps in the tutorial, the Step 1. DIA GPF / gpf_results_importer.sky file should have opened after the import was finished, or you have the gpf_results_manual.sky version made. From one of these files, launch the PRM Conductor program (a), and Skyline’s bottom bar will report the progress of creating a Skyline custom report file that gets loaded by PRM Conductor (Figure b). Note that PRM Conductor depends on .raw files for some analysis, so it will look in obvious locations for the data files referenced in the Skyline file. If the raw files were not found, it will ask you to find the missing files. Once you double-click and use the file chooser to select them, the dialog will show a green “True” and you can continue. Even if the Skyline file used .mzml or another raw file format, just place the associated .raw files in a nearby folder and you can use the PRM Conductor.
Once loaded, the user interface in the figure below will be shown. There are 3 main parts to the user interface: a set of parameters on the left, a set of transition metric plots on the top right, and a set of precursor metric plots on the bottom right. We start with the Refine Targets parameters and the associated plots. Each of the text boxes has a corresponding graph on the right. The current value in the text box is a threshold value, and is displayed with the graph as a dashed, vertical line. The title of the plot reports how many of the transitions were filtered based on the current threshold. The blue distribution in the plot is for all the transitions in the report, while the red one is after all filters have been applied. Changing the parameter in the text box and pressing Enter will update the calculations in the plots on the right.
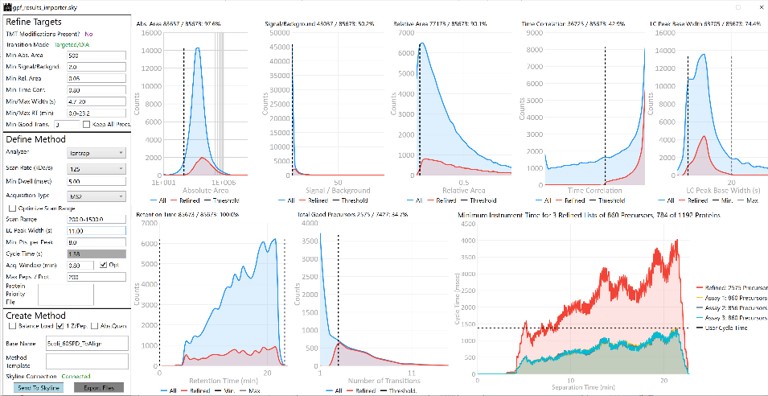
Refine Targets Section
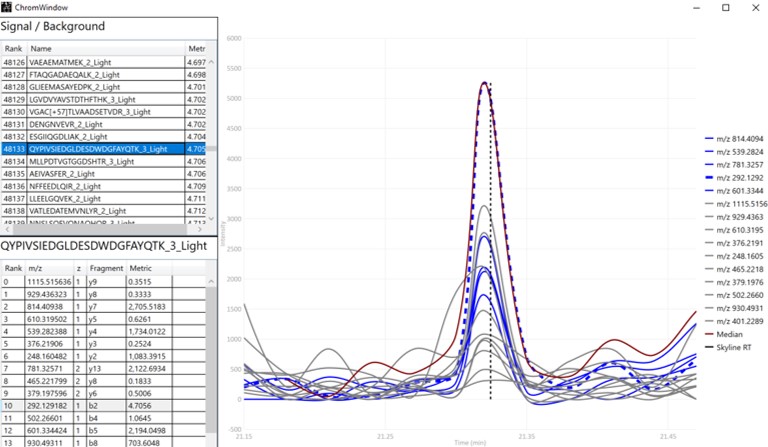
The first plot on the top is Absolute Area distribution. These are the Area values determined by Skyline for each transition. Note that these values are analyzer dependent, so a threshold used for Orbitrap discovery data may not be suitable for the Ion trap discovery data. The second plot is the Signal/Background distribution. These are the values of Area and Background as determined by Skyline for each transition. A traditional threshold for S/B is 3, however the user may wish to adjust to a different value. Highlighting the blue distribution and left-clicking will bring up a new window that gives the user an idea of what a particular S/B looks like in the Skyline transition data. For example, the figure below is a view of the plot after clicking the Signal / Background plot around 4. In the top left, all the transitions are in a grid, sorted by Signal / Background. On the bottom left is a grid showing all the transitions for the peptide corresponding to the selected transition on the top left grid. The graph on the right shows all the transitions for this peptide. The selected transition is highlighted in dashed lines. All transitions that currently meet the thresholds are colored blue, while the other transitions are colored grey. The red transition is the median transition trace.
The third plot on the top is the Relative Area distributions, that is, the area normalized to the largest transition for each precursor. The fourth plot is for the time correlation of each transition to the median transition for each precursor. The fifth plot on the top is for width of the transitions at the base, where a general rule is that outliers from this distribution may be poor quantitative markers, either because they are too narrow to characterize with the same acquisition period as wider peaks or are wide and potentially not reproducible. On the bottom row, the first plot is the retention time distributions. Precursors that are at the very beginning and very end may have the highest variability and could potentially be filtered out. The final transition filtering parameter is the Min Good Transitions text box. Many researchers prefer to set this value in the range of 3-5, to increase the confidence that a set of transitions is a unique signature for a peptide of interest. However, you could also click the Keep All Precs checkbox if you wanted to keep all the precursors, along with their best Min Good Transitions, which would potentially include some “bad” transitions. This is useful for cases with heavy standards where you don’t want to remove any of them. This is used in the Absolute Quantitation - PQ500 walkthrough.
Define Method Section
The next set of parameters on the left, in the Define Method box, control the acquisition parameters to be used in the targeted method. These parameters affect the speed of acquisition and the number of targets that can be scheduled in an analysis. The first parameter in the Define Method box is Analyzer. In this tutorial we are focused on Ion Trap analysis, but it is possible to make assays for other Analyzers. Selecting an Analyzer makes a specific set of parameters visible, which can change the characteristics of the analysis.
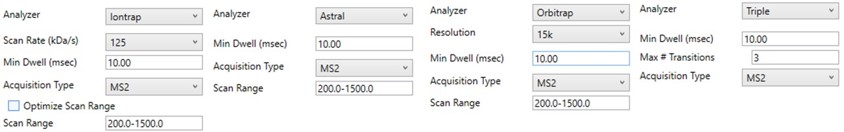
When any of these parameters are updated, the Scheduling graph on the bottom right will update. This graph shows the distribution of retention times for all Refined precursors in the red trace and the user’s chosen Cycle Time with the horizontal, dashed, black line. As we will see later, the user can choose to create a single assay that schedules as many precursors as will fit beneath that Cycle Time or may choose to create as many assays as necessary to acquire data for all refined precursors. The figure below shows views of the Scheduling graph in the single assay or “Load Balancing” mode, which we will talk more about below.
For peptide analysis, there is not much practical utility for the additional resolution afforded by the 66 kDa/s or 33 kDa/s over the “unit mass resolution” of the 125 kDa/s scan rate. Because for targeted methods we typically use the Dynamic Maximum Injection Time Mode in the instrument method file, the instrument allocates any additional time in the cycle to the targets according to their intensity. Therefore, the slower scan rates historically were useful mostly to guarantee a minimum amount of injection time per target, above the ~13 ms afforded by 125 kDa/s with the 200-1500 Th Scan Range, which can be useful for low concentration samples. With PRM Conductor, one can also set a minimum injection (or dwell time for QQQ users), which can accomplish the same thing. For peptides, we typically use the fixed 200-1500 Th scan range, because it guarantees this amount of injection time, gives a reasonable acquisition speed (~65 Hz), and the fragment ions in regions above and below this range are not essential to characterize most tryptic peptides.
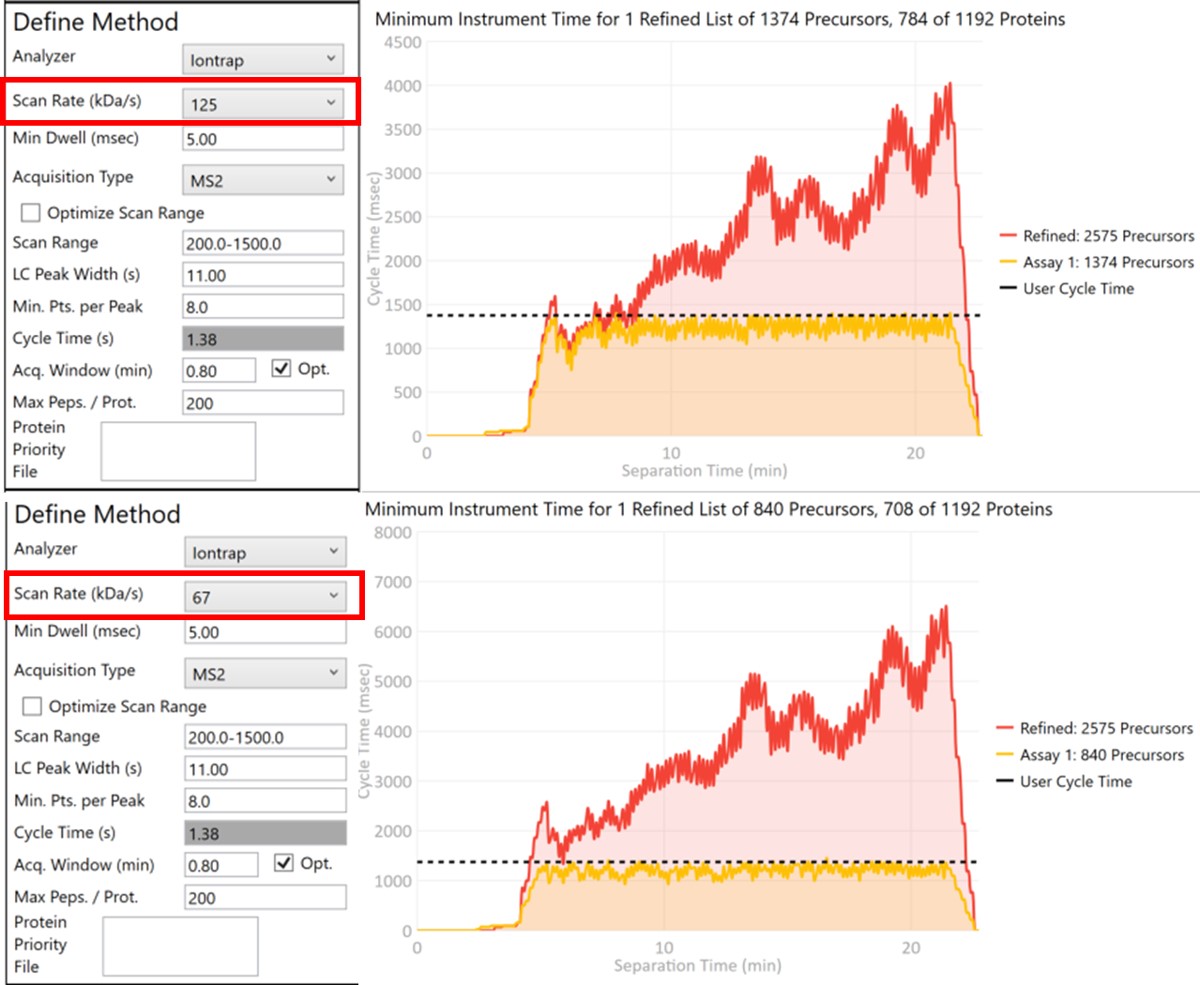
Let’s see what effect the Scan Rate has on the experiment. Changing the Scan Rate updates the estimation of how fast the instrument can acquire data. This is reflected in the Minimum Instrument Time plots, where in the top view, the Scan Rate is set to 125 kDa/s. An assay that would acquire data for all 2575 Refined Precursors would take 4 sec at the peak (at ~21 min), and 1374 precursors are able to be scheduled with the user-defined Cycle Time of 1.38 seconds. When the Scan Rate is set to 66 kDa/s (bottom part of the figure) the 2575 Refined Precursors would take 6.5 seconds of instrument time, and only 840 precursors can be scheduled with a Cycle Time of 1.38 seconds.
The user can change any of the parameters in this pane and visualize the effect on the assay. Of particular importance is the Acquisition Window. Stellar instruments are enabled with a new algorithm for real-time chromatogram alignment called Adaptive RT, which allows the instrument to adjust when targets are acquired to account for elution time drift. We find that an Acquisition Window setting of 0.75-1.00 minute usually results in good data, at least for the gradients we have typically used, in the range of 30 to 60 minutes. As LC peak width decreases for shorter gradients, we have successfully used narrower Acquisition Windows of 0.5 or even 0.3 minutes. Traditional tMS2 experiments, without real-time alignment, can sometimes require Acquisition Windows in the range of 3-5+ minutes. Adjusting this parameter allows to visualize the gain in number of targets afforded by narrower Acquisition Windows. For example, a 1 minute versus a 5 minute Acquisition Window allows to analyze more than 3x more targets (1172 versus 305). An additional feature related to Acquisition Window is the Opt check box. We have observed that the most variable part of many separations is at the beginning, which poses additional challenges for a real-time alignment algorithm because few if any compounds are eluting at the beginning. The Opt checkbox expands the Acquisition Windows somewhat, while still respecting the user’s Cycle Time requirement. The targets that are most improved by this algorithm are the early and late eluting targets; those where expanding their acquisition windows has little effect on the available injection time, but where alignment can be the trickiest.
The last two parameters, which will be explored more below, are the Max Peptides per Protein, which helps to focus the analysis on just the highest quality peptides from each protein, and the Protein Priority File, which allows to create exceptions for particular proteins using their Skyline protein names.
Create First-Draft Targeted methods and Skyline analysis file
Exporting Multiple Methods for All Refined Precursors
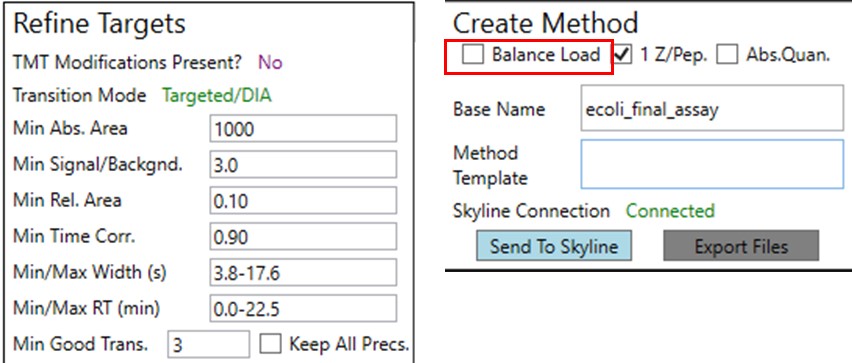
Now we will create multiple tMS2 methods for all the precursors that passed the filters and check to see which ones are stable and reproducible quantitative targets. To do this, uncheck the Balance Load button, which changes the Minimum Instrument Time plot so that all the Refined precursors are split into 3 assays, each of which require less acquisition time than the 1.38 second Cycle Time. Enter a base name for the file that will be created, otherwise the name “assay” will be used. Double-click the Method Template box and find the Step 2. Validation with Subsets/60SPD_PQ500PRTC_AlignTemplate.meth file. Then click the Export Files button.
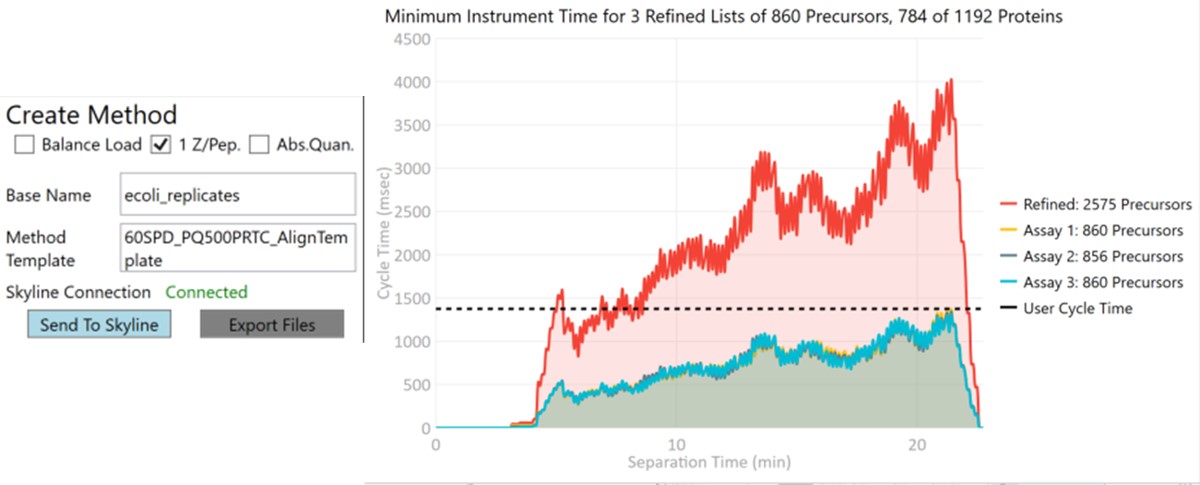
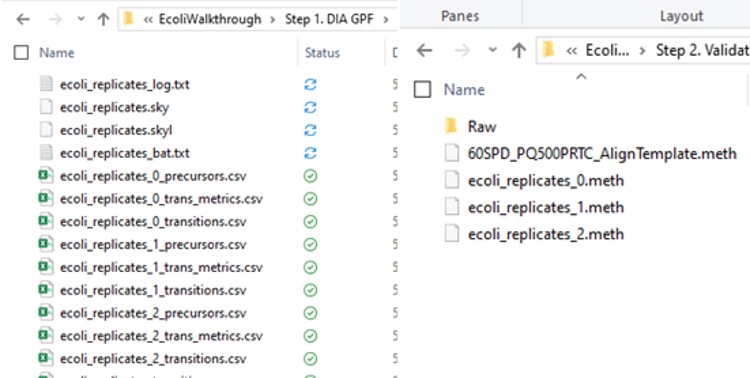
Some processing of the raw files will take place, as denoted by a progress bar, while the data files are aligned in time and a Adaptive RT reference file with the .rtbin extension is created. This processing only happens once, so if PRM Conductor is ever launched again for these raw files it won’t take as long to Export. The processing can take several minutes, depending on how many files are present and how long the gradient was. Method files will be created for each of the 3 assays in the same folder as the tempate method. A Skyline file will be created that is configured for tMS2 analysis with settings appropriate for the selected Analyzer. As a backup for the method and Skyline files, isolation and transition lists lists will be saved that are suitable for importing to the method editor or Skyline. The figure below is a view of the Step 1 folder showing some of the created files, and also the Step 2 folder showing the exported method files.
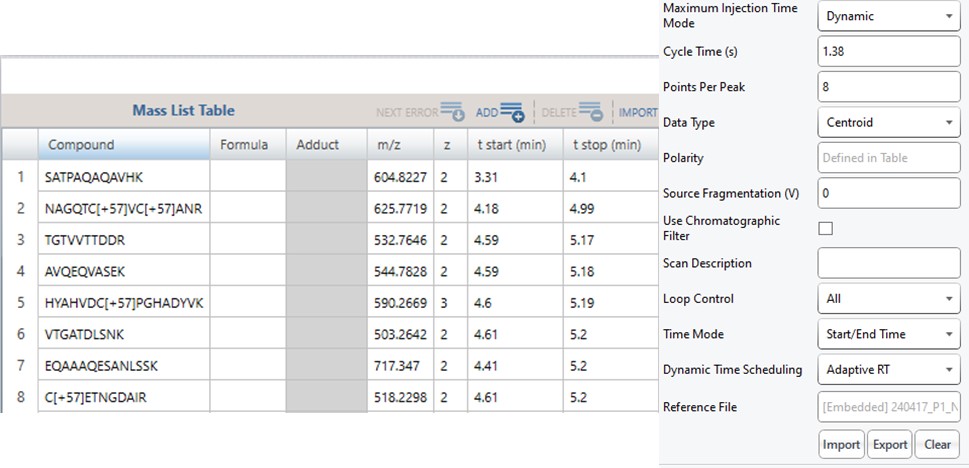
Note that because the 60SPD_PQ500PRTC_AlignTemplate.meth file had an Adaptive RT experiment and the tMSn method had Dynamic Time Scheduling set to Adaptive RT, the methods have embedded a .rtbin file to use for the real-time alignment. Notice too that the user’s Cycle Time and Points Per Peak were included, as well as the relevant precursors with their m/z, z, and scheduled acquisition times.
When exporting method files, a feature that can be useful is the Protein Priority File. There are two reasons to use this feature:
- when exporting multiple replicates, the peptides from any proteins on this list will be added to each of the replicates. The purpose of this is to support Skyline’s iRT feature, which requires the same iRT peptides to be present in each replicate. We explore this more in the walkthrough on absolute quantitation.
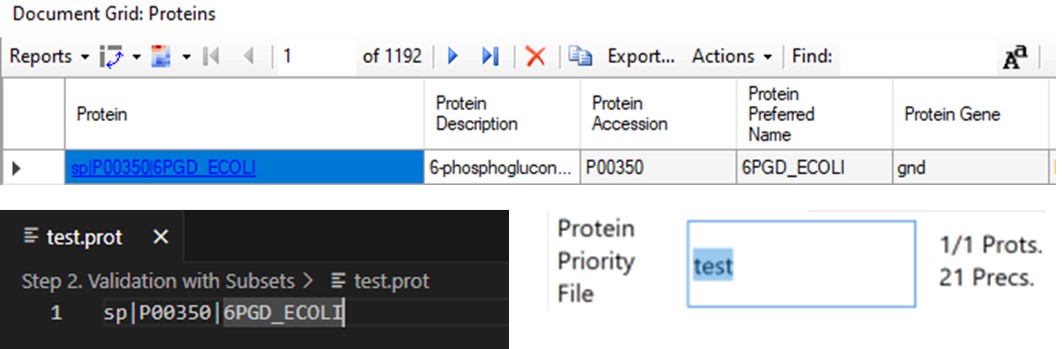
- When exporting a single replicate (balance load mode), any peptides from proteins on this list are added to the final list, assuming they passed the transition and precursor filters. To use the Protein Priority File feature, just create a text file with the .prot extension, and add any prioritized proteins to it, one per line, using exactly the format that Skyline uses for the Protein Name. For example, one could add sp|P00350|6PGD_ECOLI, as seen in the figure below in the Protein document grid, but not the Gene name displayed in the Skyline tree. When the .prot file is selected, the UI updates to display how many proteins and peptides are being prioritized. For this walkthrough we didn’t use the Protein Priority File feature, however.
Acquire First-Draft Targeted data on Stellar MS
If the user desired, they could rely on just the results from the library to create a targeted assay. However, because a targeted assay may be used for many samples, we have found it worth the extra effort to further validate the set of precursors for reproducibility by performing several injections with each of the first-draft assays and filtering on a minimum coefficient of variation (CV) on the LC Peak Areas. In some more advanced scenarios where the assay is part of a multi-proteome mixture, one could perform injections for at least two concentrations of the proteome of interest, to ensure that the selected peptides change area with concentration appropriately, and thus belong to that proteome. In this tutorial we will demonstrate filtering performed on the LC peak area CV.
Import First-Draft results to Skyline
We performed 2 injections for the 3 first-draft assays. The program was in a slightly different state at the time, so the data collected don’t perfectly match the Skyline file created in the previous step, but they are very close. Normally we could just use the ecoli_replicates.sky file created in the last step for importing the results, but because of the different program state, we'll instead do a Save As on the Step 1. DIA GPF/gpf_results_importer.sky and save a new file, Step 2. Validation with Subsets/ecoli_subset_replicates.sky.
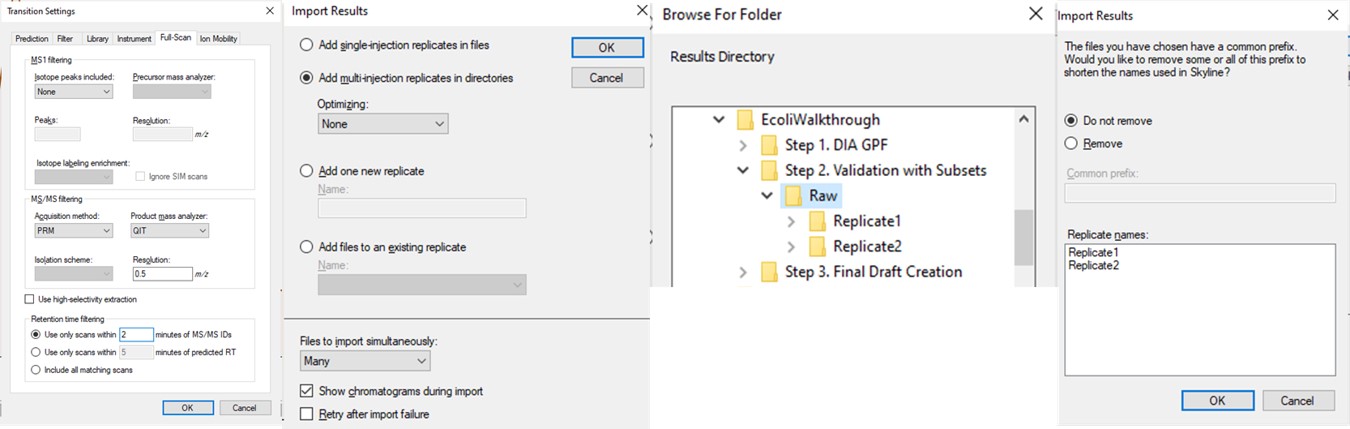
- Change the Transition Settings / Full Scan / Acquisition method to PRM with QIT analysis, and set the Retention Time filtering to a value that will probably cover the retention time shifts in the coming weeks that this assay will be run, +/- 2 minutes.
- Use Edit / Manage Results and remove the Chrom_Lib_Replicate.
- Use File / Import / Results and select Add multi-injection replicates in directories, and then choose the Step 2. Validation with Subsets\Raw folder in the dialog that pops up. Press Okay and we can also Not remove the common 'Replicate' in the folder names. Loading the data as multi-injection replicates, them nicer to view in Skyline, because each of the multiple raw files involved in a replicate is merged into a single result, and peptides won’t show up as “missing” if the wrong result file is selected.
- Now though because we started from the gpf_results_importer.sky that has all the peptides, we should Refine / Remove missing results, and then Refine / Remove empty proteins.
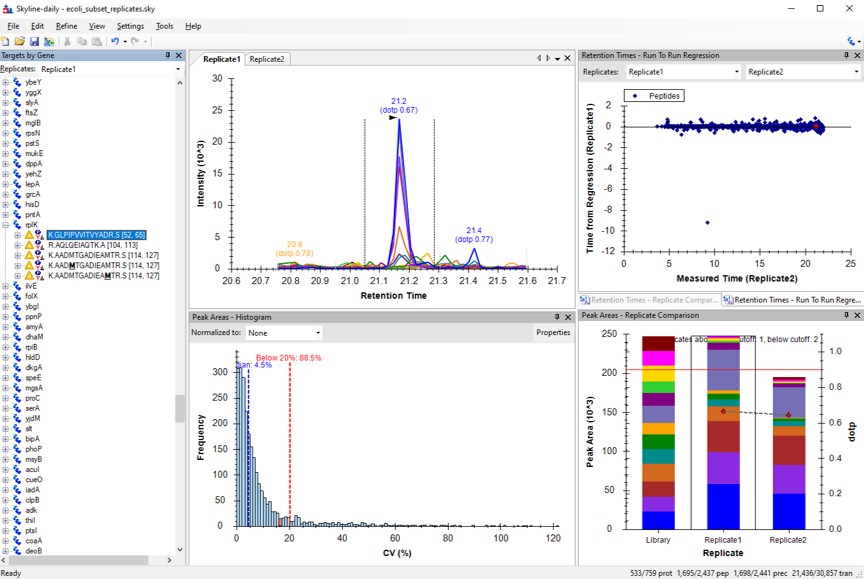
- Arrange the graphs like in the figure below.
- View / Retention Times / Replicate Comparison
- View / Retention Times / Regression / Run-to-Run
- View / Peak Areas / Replicate Comparison
- View / Peak Areas / CV Histogram
Final-Draft Creation
CV Refinement in Skyline
We have kind of a chicken-and-egg dilema now. As the .sky document currently has all of the up to 15 library transitions for each peptide, should we filter them first, and then filter the precursors by CV, or should we filter by CV and then filter the transitions? Here we opted to do the following to explore all the different options:
- Use Save As and save ecoli_subset_replicates_refined.sky. Sometimes when you save the document, the transitions go back to their original state. So it's better to save the document before you make any big changes.
- This switching of the transitions back to their original state happens if Settings / Transition Settings / Filter / Auto-select all matching transitions is selected. Go there and unselect that option, and press Okay.
- Run Tools / Thermo / PRM Conductor
- Select the Keep All Precs. option, so that we don't filter precursors, and keep a minimum of 3 transitions for each one. If there are only 2 good transitions, the 3rd one will be the next highest scoring, where the score is intensity x time_correlation.
- Press the Send to Skyline Button
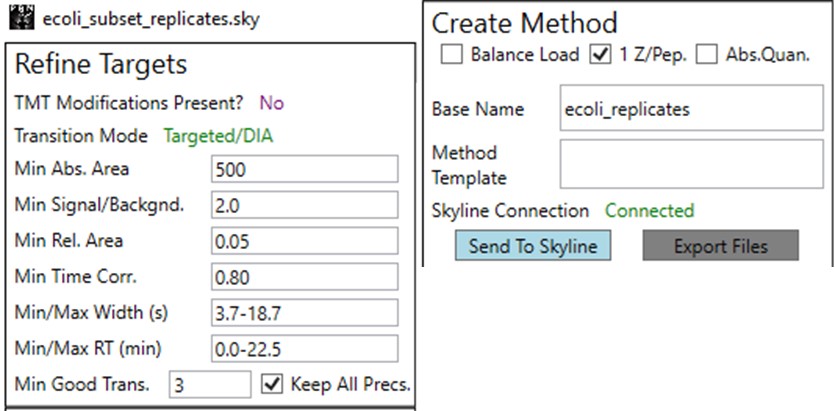
You can use the undo/redo buttons to see the effect that the filtering has had on the transitions. For example, in the figure below is shown the data for the AQLQEWIAQTK peptide before and after filtering.
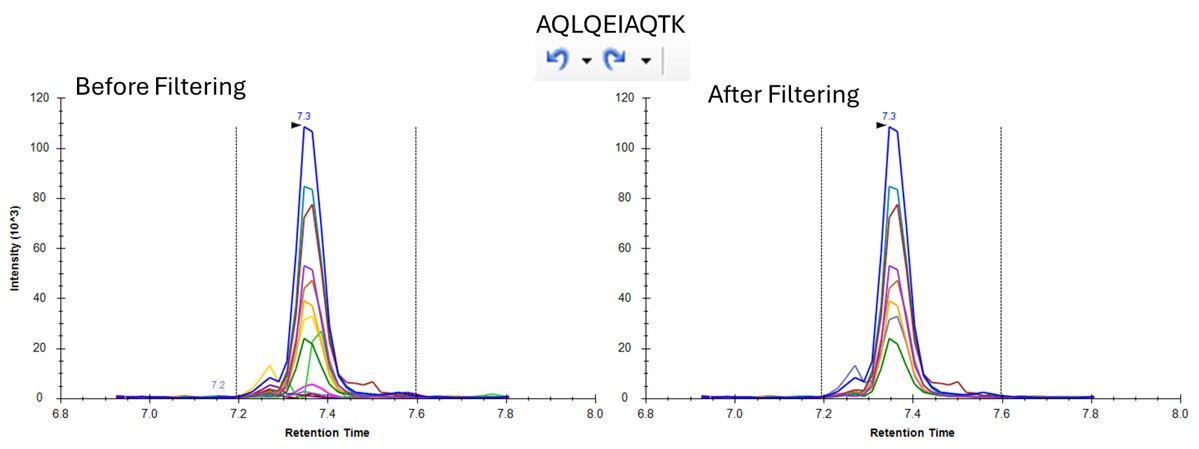
- One can click on the CV histogram, and Skyline opens a Find Results pane, that allows you to click on peptides and explore what the peaks look like at various CVs. You could also use View / Peak Areas / CV 2D Histogram to see a rather typical plot, where the most abundant peptides have the lowest CV's, in general, although bad CVs can be found at almost any intensity. Or good CVs at low intensity, for that matter.
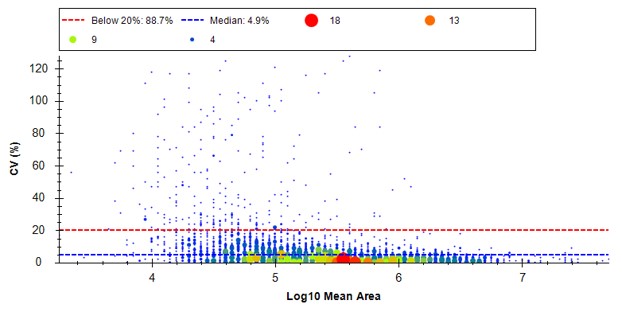
Because we have a MS experiment in our method, the Total Ion Current normalization method is used, which can help normalize out experimental variation like autosampler loading amounts.
- Save the Skyline file as ecoli_subset_replicates_refined_cv.sky.
- Use Refine / Advanced / Consistency and enter 30 in the CV cutoff %. If you were going to use TIC Normalization, then be sure to select that option in the Normalize to: combo box. Press Okay. The CV histograms will update, and the number of proteins, peptides, precursors, and transitions will update in the bottom right hand corner of the Skyline document. You can use the undo/redo to check the effect. Save the Skyline document.
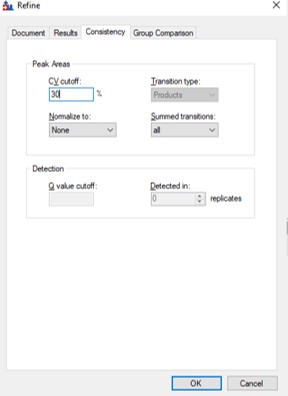
Filtering + Load Balancing
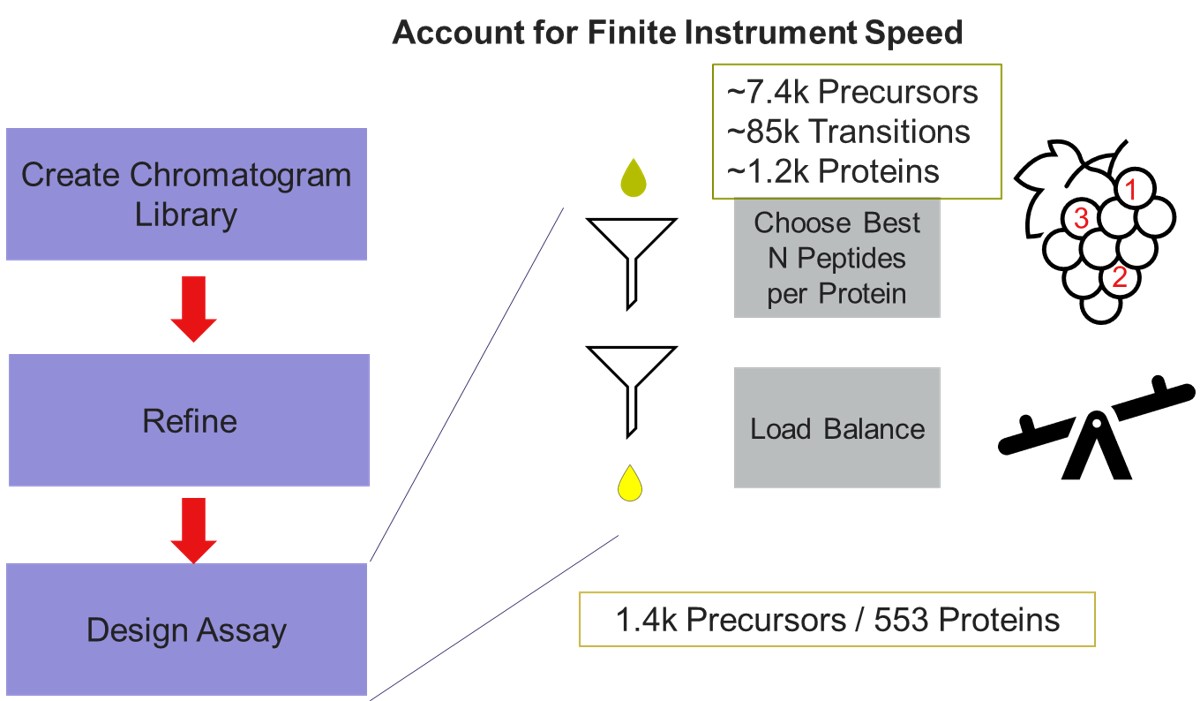
Now we will design the Final Draft assay. Our goal for this section is to ensure that the instrument can acquire quality data all the targets that are of interest. Here the experimenter can make certain concessions, such as, how many peptides per protein would give a usable result? Or am I willing to use 7 points across the peak instead of 10? We send the data to the PRM Conductor tool for analysis again using Tools/Thermo/PRM Conductor and get a View of the program like below. Compared to the earlier figure when PRM Conductor was run on the discovery results, these graphs are much different, because the precursors and transitions picked were already of such high quality. The percentage of transitions retained in the titles of each graph is close to 90% or greater. We set the LC Peak width to an even 11 seconds after hovering our mouse over the LC Peak Base Width graph to see the apex width, and set the Acquisition Window to 0.8 min.
This assay is what we would currently consider "normal", or conservative. There are 1411 precursors in a 24 minute method, which is about 3.5k precursors/hour. Had there been more precursors at earlier retention times, these settings would give about 5k precursors/hour. For fun, we have included HeLa results in the Step 1. DIA GPF\Processing\HeLaResults folder for those that wanted to explore what happens with a massive number of possible precursors.
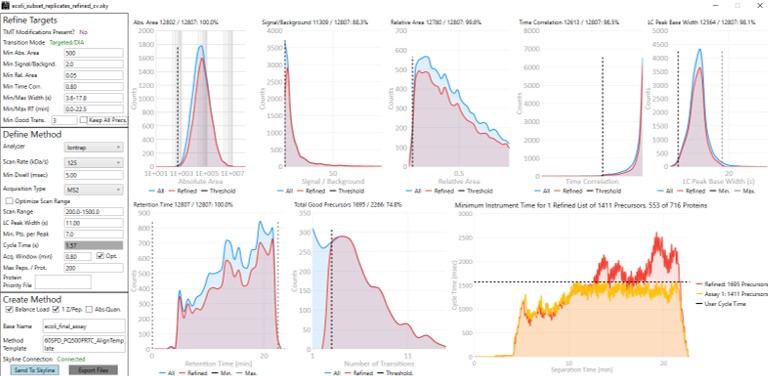
To realize the maximum throughput on Stellar MS, the user can select the Optimize Scan Range button, and could try using 6 points per peak, which is sometimes considered to be the Nyquist frequency for a Gaussian peak. We have successfully used such settings and observed little change in the results. The tradeoff being made with these settings is in the minimum amount of injection time per acquisition.
- The conservative mode guarantees about 13 milliseconds of injection time, because that's the largest injection time that doesn't slow down an HCD acquisition. If you made a method with the Maximum Injection Time mode set to Auto in the Method Editor, this is the injection time you would get. We like to use the Dynamic Maximum Injection Time Mode, where the precursors that have extra time in the their cycle, like the ones at 8 minutes, would get that extra time distributed to them.
- The aggressive mode only guarantees as much injection time as would not slow down the acquisition that has a customized scan range for the transitions that pass the PRM Conductor filters. This could be as little as the value in the Min Dwell Time box, currently set to 5 milliseconds. The rule-of-thumb is that signal-to-noise goes up (and LOQs get better) as the square root of the injection time.
- If you export a method with the Optimize Scan Range, you can see all the various, customized scan rates for each precursor.
- This mode can achieve more than 8000 peptides/hour throughputs. Play with the HeLa results to check this out.
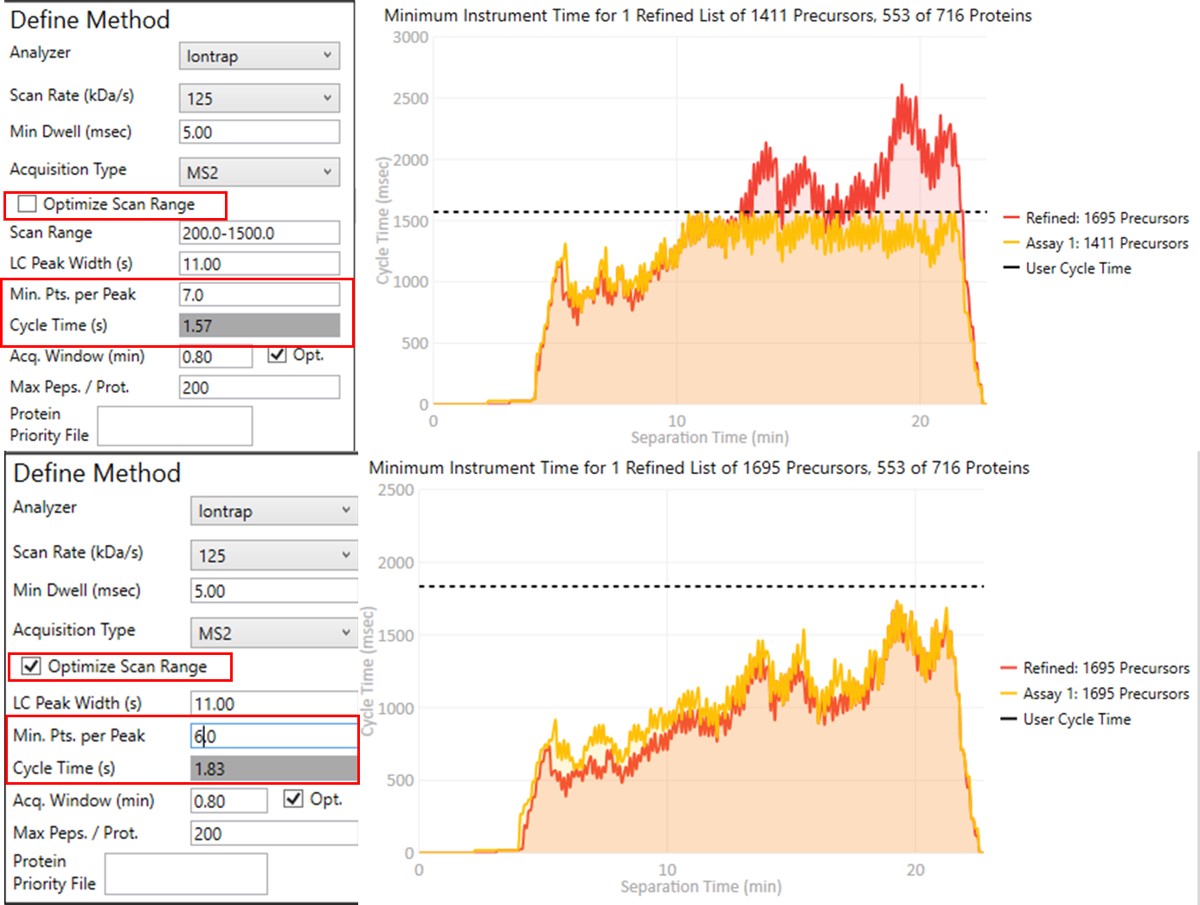
Create Final-Draft Targeted Method and Skyline file
Notice that in above figures, the Balance Load check box is checked, and so the yellow Assay 1 trace in the Minimum Instrument Time plot is flattened up against the user Cycle Time. If the Max. Peps/Prot. value was reduced from 200 to 2, then the assay would contain only 869 precursors precursors, and there would be more space between the top of the Assay 1 trace and the horizontal cycle time line. Peptides are added from each protein in order of their quality from highest to lowest, as long as there is time at that point in the assay, and as long as the protein being considered has less than Max. Peps/Prot. peptides scheduled. It’s possible that an assay with fewer targets would have better quantitative performance, due to the longer available maximum injection times, and that could be of interest to the experimenter. In this example we leave the Max Peps./Prot. value at 200.
- Click on Export Files to export a method. Once again, some processing takes place as the raw files are analyzed, and finally isolation and precursor lists, as well as method and Skyline files are created. This is the final draft assay, ready to be used for targeted experiments.
- Normally we would use either the created Skyline file to collect our next data sets, or we would Save the current Skyline file with a new name, and press Send to Skyline from PRM Conductor to update it. In this case, because as mentioned above, the subset validation assays were slightly different when the data were collected compared to this walkthrough, we'll use the Skyline file actually used for data collection in the next step.
Analysis of Final Assay Replicates
Final Assay Replicates
Many researchers are not quite done once they have a "final-assay". Typically users want to perform a larger number of replicate injections with this candidate assay. A typical practice for assays that will be used for a large number of samples is to characterize the stability of the peptides and reproducibility of the LC/MS system using a 5x5 experiment. In this experiment, 5 different samples are digested and prepared, and each aliquoted into 5 vials, for 25 total. The vials are stored in the autosampler or a refridgerator at the same temperature, and each day for 5 days in a row, at least 5 replicates are acquired for each of the 5 vials. The inter-day, intra-day, and inter-sample variability can be assessed, and poorly performing peptides can be removed.
Here we will simply load some replicate data acquired with a final assay very similar to the one that we created in Step 2 of the walkthrough.
A Note about Spectral Libraries
An action that we could have included at the end of Step 2 is to create a final spectral library, once all the final peptides are refined and their retention times are known. In the future when replicates are imported, the retention time filtering for MS/MS IDs will be relative to the spectral library you have created, and not from the discovery runs, which can be useful. We can do this from the Step 2. Validation with Subsets\ecoli_subset_replicates_refined_cv.sky file.
- Use File / Export / Spectral Library
- You could use a name like ecoli_final_assay in the dialog that pops up. Note that when we exported a final method in the last step, the script actually created that library, so you don't have to overwrite it.
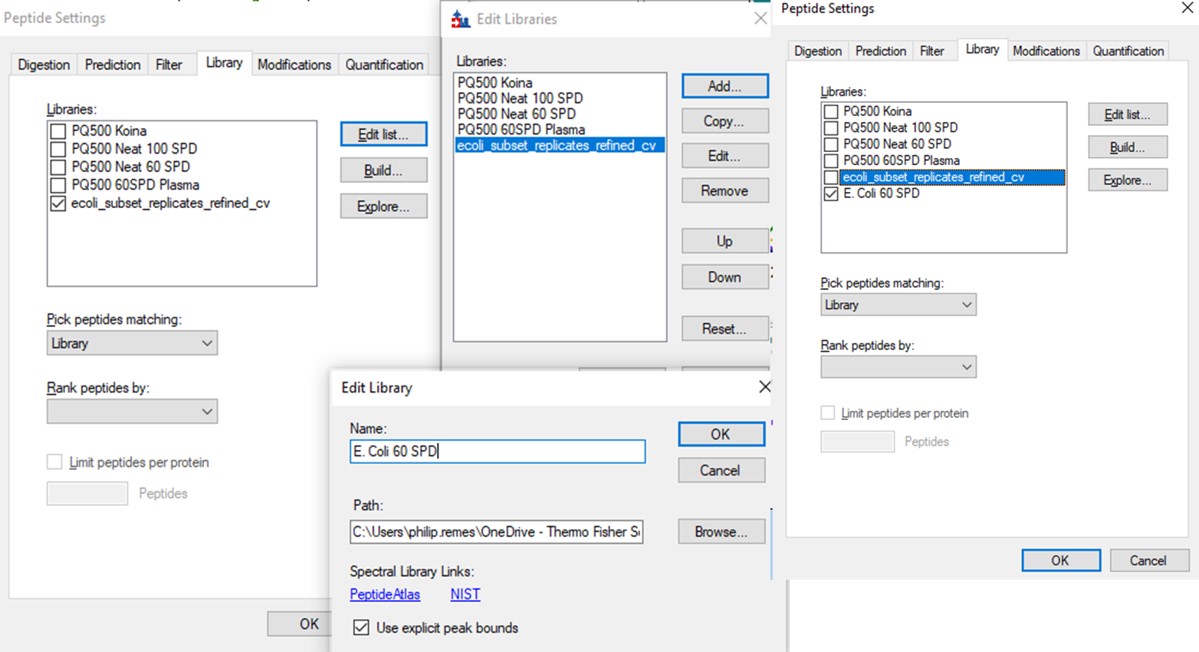
- To use the new library, you go to Settings / Peptide Settings / Library, and Edit list.
- Move the cursor to the bottom of the list, so the new library will be added there. Press Add.
- Find the location of the library you made, and give it a name.
- Then unselect the previous library, and select the new library.
- Press Okay to exit. Now the new library is being used.
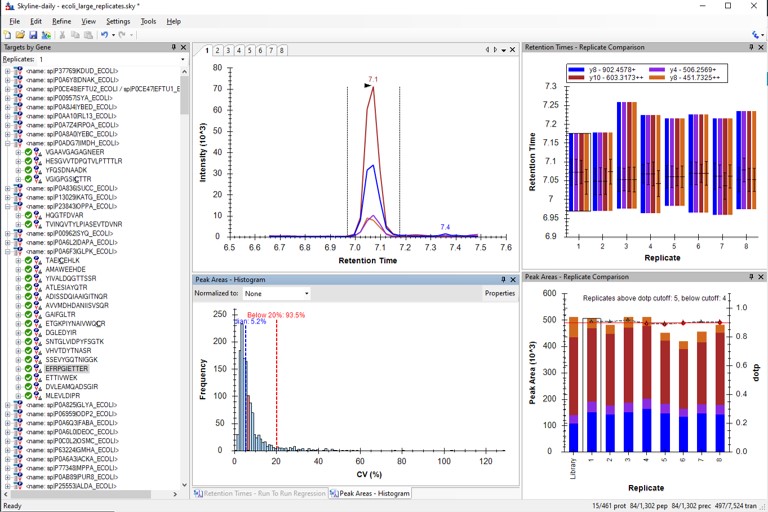
- As it is, we'll use the library that comes with the file Step 3. Final Draft Replicates\ecoli_large_replicates.sky. Open this file.
Loading and Analyzing the Results
- Use File / Import / Add single-injection replicates in files, and add the 8 files in Step 3. Final Draft Replicates\Raw.
- After these load, you can arrange the Skyline document as before.
- Save the Skyline file as ecoli_large_replicates_loaded.sky.
Use View / Retention Times / Regression / Run-to-Run. There are some peaks that are not consistently picked. Click on any of these outliers to see what they look like. Most are some kind of noise. Maybe they were "reproducible noise" in a previous step, or maybe there are interferences that get picked up over time with multiple replicates.
We could filter out the remaining peptides with poor CV's. This actually gets rid of all but a few of the retention time outliers. Additionally we could run PRM Conductor, and set some more stringent filtering settings, like in the figure below. We would unclick the Balance Load box to ensure that no precursors are filtered based on whether they fit in less than the cycle time, and press Send to Skyline.
Here we see the effect on the retention time outliers from run to run. Almost all the outliers are gone.
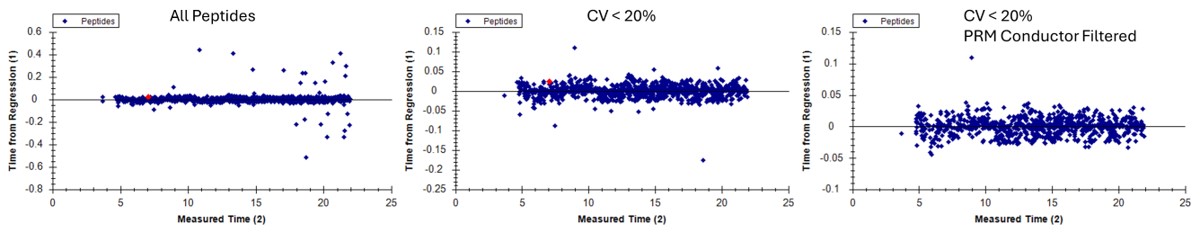
- Make sure that Settings / Transitions Setttings / Filter / Auto-select all matching transitions is not selected, and then Save As ecoli_large_replicates_loaded_refined.sky. This assay is has removed a few precursors, and could be used going forward.
Filtering Retention Time Outliers in Another Way
Let's try another way of filtering the retention time outliers. Go back and open the file Step 2. Validation with Subsets\ecoli_subset_replicates_refined_cv.sky.
- Open Settings / Peptide Settings / Prediction.
- Select Add from the Retention time predictor drop down menu.
- Select Add from the Calculator drop down menu.
- In the iRT standards drop down, select Add.
- In the Calibrate iRT Calculator, press Use Results, and enter a number like 100 in the Add Standard Peptides dialog that comes up, and press Okay.
- Give the Calculator a name like E. Coli PRM Assay and press OKay in the Calibrate iRT Calculator.
- In the Edit iRT Calculator, press Add Results, and then Press okay when asked to add these peptides, and if the standards should be updated.
- Press Create on the iRT database button, and select Yes when ased to create a new database file. Save as Step 3. Final Draft Replicates/ecoli_irt_prm.irtdb.
- In the Edit iRT Calculator dialog still, give it a name like E. Coli PRM and press Okay.
- In the Edit Retention Time Predictor, set 2 minutes, and press Okay.
- We are still in the ecoli_subset_replicates_refined_cv.sky file, so we don't need to use the calculator here. Set None in the Retention time predictor, press Cancel, and close this Skyline file.
- Open up our ecoli_large_replicates_loaded.sky file, where we have all the retention time outliers.
- Go to Settings / Peptide Settings / Prediction and the E. Coli PRM name should be there. If it wasn't, for some reason, you could add the calculator we created from the Edit List option. Select the E. Coli PRM calculator.
- We were asked to add some standard peptides, because some of the ones that went into the calculator are not present. You can add them or not add them.
The background of the chromatogram plots is now beige, and there is a faint vertical line that says "Predicted" on the plots, like below.
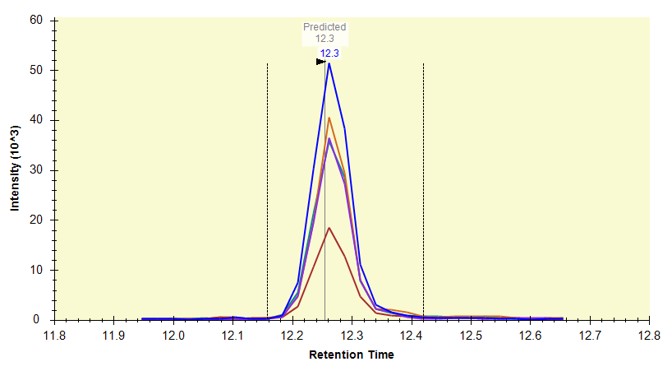
Use View / Retention Times / Regression / Score-to-Run, and on the plot that comes up, right click and in the Calculator menu, select the E. Coli PRM. Make sure that you are in the right-click, Plot / Residuals mode. Some of the dots in the plot are pink just because our document here has some peptides that weren't in the calculator. That's fine for this walkthrough.
- Use Refine / Advanced / Results and set Retention time outliers to 0.999 and press Okay. These peptides that were pink dots are now removed.
- You can do the same thing by right clicking the plot and setting the threshold, and then right clicking again and selecting Remove outliers.
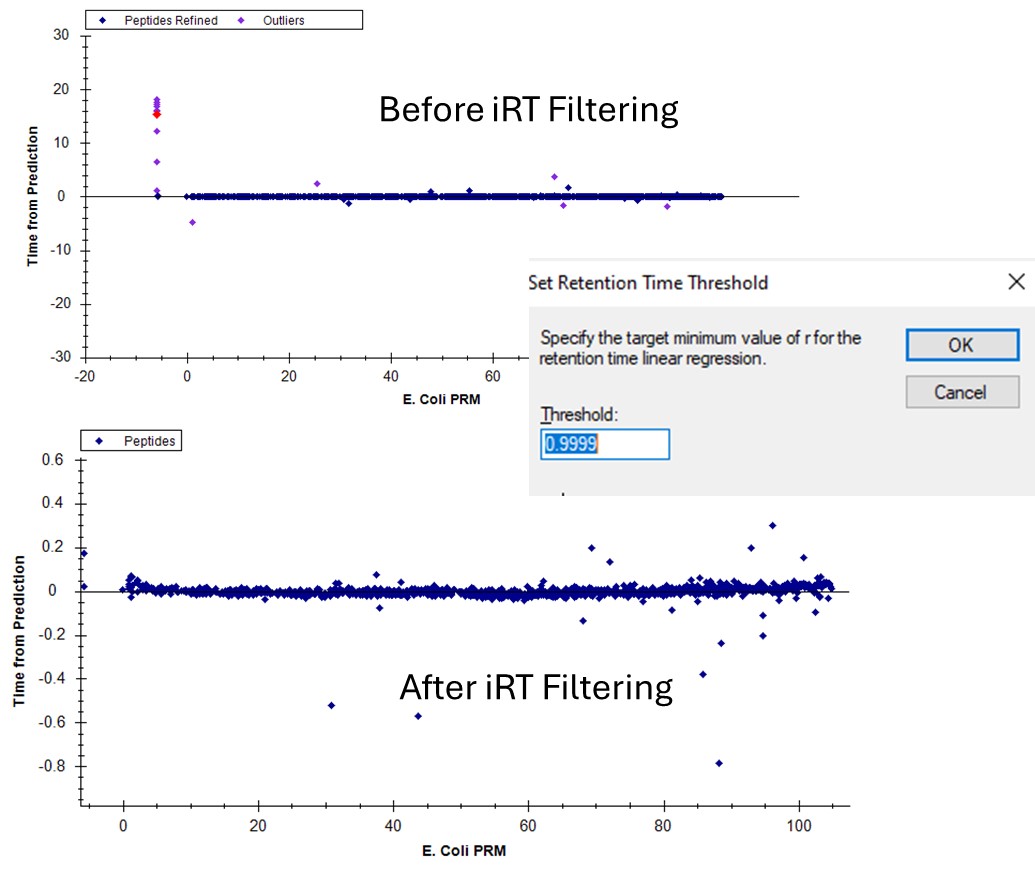
If we go back to the Run-to-Run regression, there are still a few outliers. Apparently we can't yet filter based on the experimental Run-to-Run deviations, but maybe in the future. This iRT filtering maybe wasn't as powerful in this example as just using the CV and PRM Conductor filtering, but it's another tool in our belt.
You've reached the end of this tutorial. Hopefully have a good idea of how to create an assay based on Stellar MS discovery results.